0 Genotype By Sequencing
Genotype by Sequencing (GBS): is a molecular technique that allows genomewide genotyping of a population of organisms by performing reduced representation sequencing of their genome. GBS is a robust, simple and affordable means of SNP discovery and mapping. Users of GBS often aim to find Single Nucleotide Polymorphisms (SNPs) within a population to: promote understanding of the relationships between the organisms within, or utilize the genotyping information for genomic selection, prediction or trait analysis.
0.1 Methods
Generating a reduced representation of a genome requires using restriction endonucleases to cleave the double stranded genomic DNA at any position the enzymes unique cut site is found. By utilizing one or more restriction endonucleases, the genomic DNA can be cleaved into manageable fragments. Utilizing different restriction enzymes or combinations of restriction enzyme(s) allow a GBS user to sequence different sites throughout the genome. After enzymatic cleavage the sticky ends of DNA that are left freely floating are used as anchors for the ligation of barcoded adaptors.
The barcoded adaptors serve several important functions:
- A way to indentify each sample uniquely from the others in pooled DNA sequencing
- A DNA template to use for PCR amplification of the DNA before sequencing
- A structural template to adhere other sequencing instrument specific adaptors to
After barcode ligation the DNA is amplified via PCR, purified and quantified before sequencing. Upon sequencing, generally many samples are pooled into a sequencing lane to provide cost effective sharing of the massive sequencing depth provided by todays sequencing instruments.
Your sequencing data has been produced on our NovaSeq 6000. Information about sequencing density, speed and accuracy for this instrument can be found here.
1 Sample Definitions
1.1 Input Data
Specific information pertaining to the input data for your project is shown in Table 1 below.
| Date: | 01/09/2020 |
|---|---|
| Raw Data Path: | /mnt/grl/brc/data/Client/0190620/V2 |
| Genome Path: | /mnt/genomes/Spinacia_oleracea/primary.fa |
| Enzyme: | ApeKI |
The deliverable directory contains numerous files consisting of alignment maps, variant calls, and quality control metrics. These files contain the core information from your project. Content description and explanations of these files are given in the appropriate sections below. Many of these files contain metadata which contain additional information which we do not explicitly pull into this report. Information such as the sequencing flowcell, instrument ID, and cluster are embedded in the names of the sequence alignments.
1.2 Fastq Format
Your raw GBS data can be found in the file in the FASTQ format. The Illumina file naming structure is traditionally:
{Sample_Identifier}_{Sample_Number}_{Lane}_{Read_1_or_Read_2}_001.fastq.gz
A random sample might be named:
Plate-2-Maize_S2_L004_R1_001.fastq.gz
After decompression, FASTQ files have four lines per sequence:
- Line 1: begins with a '@' character and is followed by a sequence identifier and an optional description.
- Line 2: is the raw sequence letters.
- Line 3: begins with a '+' character and is optionally followed by the same sequence identifier (and any description) again.
- Line 4: encodes the quality values for the sequence in Line 2, and must contain the same number of symbols as letters in the sequence.
For Illumina FASTQ files the sequence identifier contains information that can be used to identify metadata about the run.
@<instrument>:<run number>:<flowcell ID>:<lane>:<tile>:<x-pos>:<y-pos> <read>:<is filtered>:<control number>:<sample number>
| Element | Requirements | Description |
|---|---|---|
| @ | @ | Each sequence identifier line starts with @ |
| <instrument> | Characters allowed: a–z, A–Z, 0–9, _ | Instrument ID |
| <run number> | Numerical | Run number on instrument |
| <flowcell ID> | Characters allowed: a–z, A–Z, 0–9 | A Unique string of characters that identifies the flowcell. |
| <lane> | Numerical | Lane number |
| <tile> | Numerical | Tile number |
| <x_pos> | Numerical | X coordinate of cluster |
| <y_pos> | Numerical | Y coordinate of cluster |
| <read> | Numerical | Read number. 1 can be single read or Read 2 of paired-end |
| <is filtered> | Y or N | Y if the read is filtered (did not pass), N otherwise |
| <control number> | Numerical | 0 when none of the control bits are on, otherwise it is an even number. |
| <sample number> | Numerical | Sample number from sample sheet |
2 Quality Control
Raw sequence data may contain residual sequence that is artificially introduced as a result of the laboratory methods used to prepare the DNA for sequencing. These sequence artifacts may introduce bias an increase the complexity of downstream data processing. Additionally, sequencing quality can be affected by various factors such as variation in chemical reagents, instrument platform, temperature or humidity changes, initial DNA quality or concentration. Due to the imperfect nature of the sequencing process and limitations of the optical instruments (batch effects, sequencing error, GC bias, duplicate reads, copy number variation, mapability), base calling has inherent uncertainty associated with it. The magnitude of uncertainty in each base call is represented by an error probability or Phred (pronounced like “fred”) score. This parameter is denoted formally as \(Q\) and is proportional to the probability \(p\) that a base call is incorrect, where \(Q = -10log~10~(p)\).
Any biological inference or conclusion depends on the overall quality of your data. Therfore a quality control (QC) step is essential for providing high quality results. The following QC metrics calculated from your data are designed to help you evaluate the overall technical quality of your experiment. You should always review these metrics to ensure your data are of good quality and lack obvious problems or biases, which may affect how you can ultimately use these data.
2.1 Read Trimming
During sequencing library construction, short oligonucleotides are ligated to the ends DNA fragments to be sequenced, so that they can be combined with primers for PCR amplification. While sequencing the fragments, if the read length is greater than that of the target DNA, the adapter sequence next to the unknown DNA sequence of interest is also sequenced, sometimes only partially. To recover the true target DNA sequence, it is required to identify the adapter sequence and remove it. Moreover, to improve further the quality of the sequence reads, skewer also trims the 3’ end of the fragment until a Phred quality of 20 is reached. This is equivalent to a base call accuracy of 99%. More information regarding Phred scores can be obtained from (Ewing et al. 1998) and in (Ewing & Green 1998).
The Bioinformatics Resource Center performs a quality control step to remove any sequencing adapters and low quality bases. The trimming software skewer (Jiang et al. 2014) was used to pre-process raw fastq files. Skewer implements an efficient dynamic programming algorithm designed to remove adapters and/or primers from sequence reads. Reads that are trimmed too short to be of usable length are discarded from downstream analysis along with the corresponding read pair, in the case of paired-end sequencing.
3 Tassel GBS Pipeline Version 2
The Tassel Version 2 GBS pipeline is an extension of the Java program Tassel. Tassel GBS is a scalable high throughput GBS data analysis platform with moderate computing resource requirements (Glaubitz et al. 2014).
The new pipeline stores data to an embedded SQLite database. All steps of the pipeline either read from or write to this database. It is initially created in the GBSSeqToTagDBPlugin step. When running this pipeline, each subsequent step utilizes/adds data from the database created in the first step. A diagram of the database is presented below.
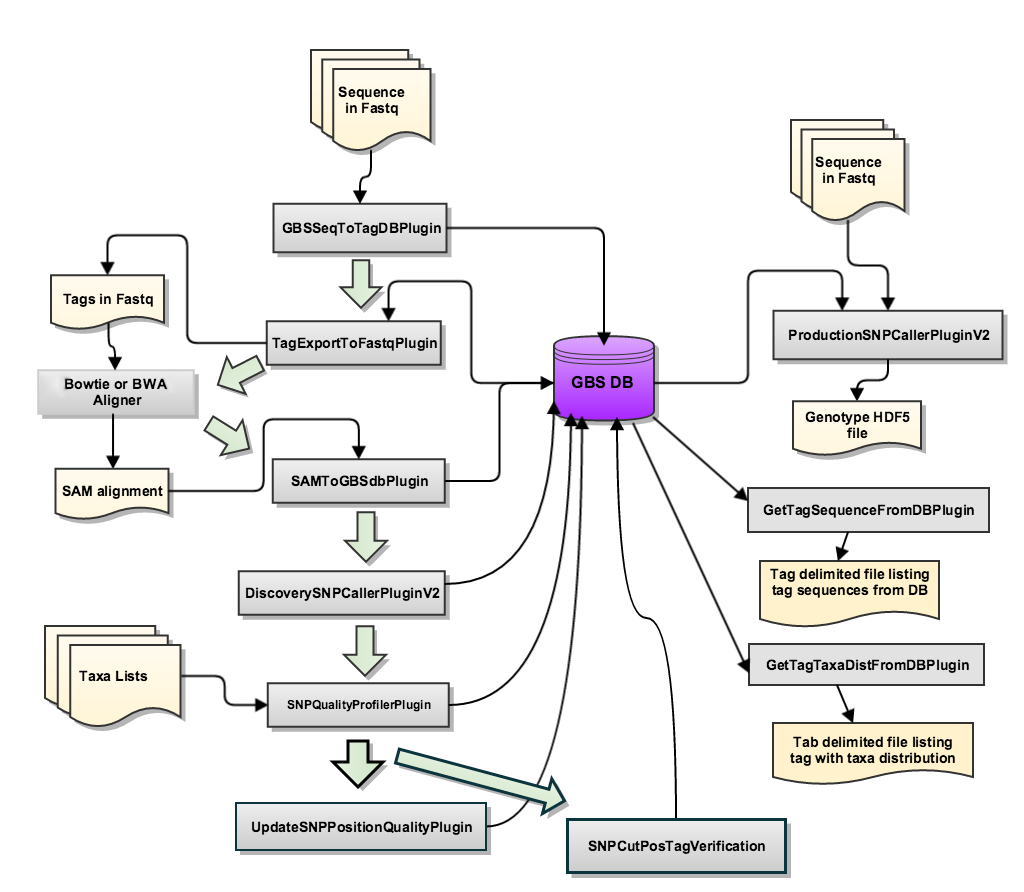
image credit: Tassel GBS v2 Bitbucket
3.1 GBSSeqtoTagDBPlugin
GBSSeqToTagDBPlugin takes fastq files as input, identifies tags and the taxa in which they appear, and stores this data to a local database. It keeps only good reads having a barcode and a cut site and no N’s in the useful part of the sequence. It trims off the barcodes and truncates sequences that (1) have a second cut site, or (2) read into the common adapter.
See the table below for a list of your input files and their associated raw and good barcoded reads:
| Fastq File | Total Raw Reads | Good Barcoded Reads | % Good Barcoded Reads |
|---|---|---|---|
| BHKT72DMXXS24_2_fastq.gz | 328727547 | 314900055 | 95.794 |
| BHKT72DMXXS25_2_fastq.gz | 346133972 | 337261358 | 97.437 |
Raw Reads: is the unprocessed reads within the file before any analysis.
Good Barcoded Reads: are sequence reads with a perfect match to one of the barcodes provided in a barcode key file and with no N’s in the sequence following the barcode up to the trim length.
See the table below to determine the read representation per sample:
| Sample Name | Demultiplexed Reads |
|---|---|
| TB01:201906241500210 | 4871283 |
| TB02:201906241500211 | 3840882 |
| TB03:201906241500212 | 3141617 |
| TB04:201906241500213 | 1220516 |
| TB05:201906241500214 | 403584 |
| TB06:201906241500215 | 3332922 |
| TB07:201906241500216 | 3356085 |
| TB09:201906241500218 | 3206050 |
| TB10:201906241500219 | 1763673 |
| TB11:2019062415002110 | 2537969 |
| TB12:2019062415002111 | 3643434 |
| TB13:2019062415002112 | 904588 |
| TB14:2019062415002113 | 1922513 |
| TB15:2019062415002114 | 3311405 |
| TB16:2019062415002115 | 2159178 |
| TB17:2019062415002116 | 3736498 |
| TB18:2019062415002117 | 3036391 |
| TB19:2019062415002118 | 1038867 |
| TB20:2019062415002119 | 4383077 |
| TB21:2019062415002120 | 3541881 |
| TB22:2019062415002121 | 2879401 |
| TB23:2019062415002122 | 1966082 |
| TB24:2019062415002123 | 2645742 |
| TB25:2019062415002124 | 1720659 |
| TB269:2019062415002163 | 3996310 |
| TB26:2019062415002125 | 2093917 |
| TB27:2019062415002126 | 4019942 |
| TB281:2019062415002161 | 4297906 |
| TB287:2019062415002142 | 4301044 |
| TB288:2019062415002143 | 1394822 |
| TB28:2019062415002127 | 31361 |
| TB292:2019062415002144 | 4693220 |
| TB293:2019062415002145 | 7144642 |
| TB29:2019062415002128 | 2095624 |
| TB30:2019062415002129 | 1473205 |
| TB311:2019062415002164 | 3242522 |
| TB312:2019062415002165 | 3942637 |
| TB313:2019062415002166 | 4693147 |
| TB314:2019062415002167 | 2697100 |
| TB315:2019062415002168 | 4542589 |
| TB317:2019062415002169 | 1417591 |
| TB318:2019062415002170 | 1249588 |
| TB319:2019062415002171 | 3262468 |
| TB31:2019062415002130 | 1319527 |
| TB320:2019062415002191 | 1927806 |
| TB321:2019062415002172 | 3336375 |
| TB322:2019062415002173 | 4053412 |
| TB323:2019062415002190 | 3570 |
| TB324:2019062415002174 | 2941917 |
| TB32:2019062415002131 | 1803268 |
| TB33:2019062415002132 | 3090496 |
| TB342:2019062415002146 | 3898621 |
| TB344:2019062415002147 | 3892773 |
| TB34:2019062415002133 | 2527338 |
| TB351:2019062415002148 | 3546380 |
| TB352:2019062415002149 | 6602342 |
| TB355:2019062415002192 | 3543571 |
| TB356:2019062415002189 | 1076192 |
| TB359:2019062415002175 | 3780938 |
| TB35:2019062415002134 | 2861343 |
| TB360:2019062415002176 | 3531495 |
| TB361:2019062415002177 | 4536715 |
| TB362:2019062415002178 | 3845707 |
| TB363:2019062415002179 | 4377290 |
| TB364:2019062415002180 | 2443930 |
| TB365:2019062415002181 | 3295198 |
| TB366:2019062415002182 | 5336955 |
| TB367:2019062415002183 | 1017752 |
| TB368:2019062415002184 | 2119291 |
| TB369:2019062415002185 | 3487084 |
| TB36:2019062415002135 | 3289158 |
| TB371:2019062415002186 | 2763307 |
| TB375:2019062415002194 | 3362717 |
| TB37:2019062415002136 | 5446743 |
| TB382:2019062415002193 | 2024661 |
| TB385:2019062415002187 | 2118758 |
| TB386:2019062415002188 | 2522771 |
| TB387:2019062415002150 | 3041348 |
| TB388:2019062415002151 | 4596016 |
| TB389:2019062415002152 | 3734361 |
| TB38:2019062415002137 | 3180931 |
| TB390:2019062415002153 | 5009953 |
| TB391:2019062415002154 | 2882213 |
| TB392:2019062415002155 | 3993264 |
| TB39:2019062415002138 | 3939640 |
| TB400:201906241500210 | 3297436 |
| TB401:201906241500211 | 2450620 |
| TB402:201906241500212 | 2269217 |
| TB403:201906241500213 | 2043302 |
| TB404:201906241500214 | 1934339 |
| TB405:201906241500215 | 1620578 |
| TB406:201906241500216 | 3072910 |
| TB407:201906241500217 | 3949526 |
| TB408:201906241500218 | 2728633 |
| TB409:201906241500219 | 3019801 |
| TB40:2019062415002139 | 5146874 |
| TB410:2019062415002110 | 3293726 |
| TB411:2019062415002111 | 1875492 |
| TB412:2019062415002112 | 3249439 |
| TB413:2019062415002113 | 2913555 |
| TB414:2019062415002114 | 2071057 |
| TB415:2019062415002115 | 1421149 |
| TB416:2019062415002116 | 2978207 |
| TB417:2019062415002117 | 2778422 |
| TB418:2019062415002118 | 3286214 |
| TB419:2019062415002119 | 3634284 |
| TB41:2019062415002140 | 3604561 |
| TB420:2019062415002120 | 3569161 |
| TB421:2019062415002121 | 2395300 |
| TB422:2019062415002122 | 3024827 |
| TB423:2019062415002123 | 2770340 |
| TB424:2019062415002156 | 6907389 |
| TB425:2019062415002157 | 3920483 |
| TB427:2019062415002158 | 5905164 |
| TB428:2019062415002124 | 2675804 |
| TB429:2019062415002125 | 4837039 |
| TB42:2019062415002141 | 3598196 |
| TB430:2019062415002126 | 4358571 |
| TB431:2019062415002127 | 2892719 |
| TB432:2019062415002128 | 2722583 |
| TB433:2019062415002129 | 2702163 |
| TB434:2019062415002130 | 2393586 |
| TB435:2019062415002131 | 2061431 |
| TB437:2019062415002132 | 2331902 |
| TB438:2019062415002133 | 3008786 |
| TB439:2019062415002134 | 558757 |
| TB43:2019062415002142 | 4395619 |
| TB440:2019062415002135 | 3902577 |
| TB443:2019062415002136 | 6149376 |
| TB444:2019062415002137 | 4628001 |
| TB445:2019062415002138 | 2960481 |
| TB446:2019062415002139 | 5211966 |
| TB447:2019062415002140 | 2340850 |
| TB448:2019062415002141 | 4919473 |
| TB44:2019062415002143 | 3661816 |
| TB451:2019062415002195 | 1051117 |
| TB453:2019062415002159 | 4219761 |
| TB454:2019062415002160 | 4092276 |
| TB45:2019062415002144 | 4168867 |
| TB46:2019062415002145 | 3281510 |
| TB47:2019062415002146 | 3998183 |
| TB48:2019062415002147 | 3264880 |
| TB49:2019062415002148 | 3983888 |
| TB50:2019062415002149 | 5438900 |
| TB51:2019062415002150 | 4072843 |
| TB52:2019062415002151 | 3902759 |
| TB53:2019062415002152 | 4558727 |
| TB54:2019062415002153 | 4345295 |
| TB55:2019062415002154 | 4012202 |
| TB56:2019062415002155 | 2905396 |
| TB57:2019062415002156 | 3934828 |
| TB58:2019062415002157 | 1416179 |
| TB59:2019062415002158 | 6208739 |
| TB60:2019062415002159 | 2530028 |
| TB61:2019062415002160 | 4539002 |
| TB62:2019062415002161 | 4370473 |
| TB63:2019062415002162 | 4066966 |
| TB64:2019062415002163 | 4761181 |
| TB65:2019062415002164 | 3230965 |
| TB66:2019062415002165 | 3967260 |
| TB67:2019062415002166 | 2896394 |
| TB68:2019062415002167 | 3316771 |
| TB69:2019062415002168 | 1153914 |
| TB70:2019062415002169 | 2232678 |
| TB71:2019062415002170 | 2040634 |
| TB72:2019062415002171 | 2796892 |
| TB73:2019062415002172 | 2094934 |
| TB74:2019062415002173 | 3095355 |
| TB75:2019062415002174 | 3640028 |
| TB76:2019062415002175 | 887970 |
| TB77:2019062415002176 | 1954851 |
| TB78:2019062415002177 | 4320311 |
| TB79:2019062415002178 | 3946669 |
| TB80:2019062415002179 | 3703259 |
| TB81:2019062415002180 | 2535635 |
| TB82:2019062415002181 | 2804720 |
| TB83:2019062415002182 | 3994439 |
| TB84:2019062415002183 | 1980138 |
| TB85:2019062415002184 | 2678242 |
| TB86:2019062415002185 | 3340099 |
| TB87:2019062415002186 | 3009971 |
| TB88:2019062415002187 | 3732187 |
| TB89:2019062415002188 | 3138272 |
| TB90:2019062415002189 | 4309035 |
| TB91:2019062415002190 | 2754301 |
| TB92:2019062415002191 | 2325574 |
| TB93:2019062415002192 | 4015065 |
| TB94:2019062415002193 | 2046390 |
| TB95:2019062415002194 | 3085905 |
| TB96:2019062415002195 | 1710394 |
| blank2:2019062415002162 | 254 |
| blank:201906241500217 | 90212 |
| Total: | 606654479 |
Demultiplexed Reads are sequence reads with a perfect match to one of the barcodes provided in a barcode key file and with no N’s in the sequence following the barcode up to the trim length.
Interpretation: The list above provides the Demultiplexed Reads assigned to each sample. Therefore, the table values should provide a clue as to which samples are most well represented in your GBS experiment (>1x106 Demultiplexed Reads). Low counts (i.e. <1x105) likely indicates sample dropout. Sample dropout occasionally occurs in GBS due to the large number of samples that are pooled on our patterned Illumina flow cells and the error inherent in measuring DNA libraries.
3.2 TagExportToFastqPlugin
TagExportToFastqPlugin retrieves distinct tags stored in the database and reformats them to a FASTQ file that can be read by the Bowtie 2 or BWA aligner program. This plugin leads to one of the main practical advantages of the Tassel GBS pipeline. Namely, the collapse of redundant reads into tags of a predefined tag size. This read collapse drastically shortens the time required for tag/read alignment because of the reduced number of tags compared to the input reads. We utilized 64 bp tags during the analysis of your data. Even a single bp difference in reads of similar sequence will lead to different tags being stored in the database. The output file of TagExportToFastqPlugin is the input to the DNA sequence aligner Bowtie 2.
3.3 GBS Tag Alignment
The BRC uses the Bowtie 2 alignment software for alignment of the FASTQ data
transfered from the TagExportToFastqPlugin. Bowtie 2 is a fast and memory efficient read aligner that supports gapped, local and paired-end alignment modes (Langmead et al. 2012) .
To narrow down the locations a read can map in a large genome, Bowtie 2 extracts multiple "seeds" (smaller DNA sequences) from each
read/tag and aligns them in an ungapped manner to the FM index in an effort to align the entire read. This multiseed heuristic
increases the speed of alignment but can reduce accuracy.
Bowtie 2 deals with inaccuracy of read mapping via two methods:
- Using an alignment scoring scheme which penalizes inaccurate alignments (gaps,insertions and mismatches relative to the reference) and rewards accuracy (correctly matched bases). Simply put, the higher the score the more similar the read/tag is to the reference sequence. If a read's alignment score exceeds the minimum score threshold (adjustable), the read's alignment is considered "valid". The scoring scheme is implemented differently for local vs end-to-end (default) alignments. In the Tassel v2 GBS pipeline we employ the --very-sensitive-local method during mapping of tags to the genome to ensure high accuracy of each tags mapping location.
- Outputting a Mapping Quality (MAPQ) for each read alignment. The MAPQ in Bowtie 2 represents the aligners confidence that the location the read is aligned to is the read's true point of origin. MAPQ ranges from (lowest: 0 --> highest: 42) and is reported as: Q = -10 log10 p, where p is an estimate of the probability that the alignment does not correspond to the read’s true point of origin.
The output of each Bowtie 2 run is a Sequence Alignment Map (SAM) file, the details of which can be found below:
| Col | Field | Description |
|---|---|---|
| 1 | QNAME | Query (pair) NAME |
| 2 | FLAG | bitwise FLAG |
| 3 | RNAME | Reference sequence NAME |
| 4 | POS | 1-based leftmost POSition/coordinate of clipped sequence |
| 5 | MAPQ | MAPping Quality (Phred-scaled) |
| 6 | CIGAR | extended CIGAR string |
| 7 | MRNM | Mate Reference sequence NaMe (‘=’ if same as RNAME) |
| 8 | MPOS | 1-based Mate POSition |
| 9 | ISIZE | Inferred insert SIZE |
| 10 | SEQ | query SEQuence on the same strand as the reference |
| 11 | QUAL | query QUALity (ASCII-33 gives the Phred base quality) |
| 12 | OPT | variable OPTional fields in the format TAG:VTYPE:VALUE |
This data has been provided to you in the GBS_
See below for a table indicating genome mapping of the tags in your population:
| Total Tags | Secondary Tag Alignment | % Secondary Tag Alignment | Supplementary Tag Alignment | % Supplementary Tag Alignment | Mapped Tags | % Mapped Tags |
|---|---|---|---|---|---|---|
| 1175237 | 0 | 0.0 | 0 | 0.0 | 599318 | 50.996 |
3.4 SamToGBSdbPlugin
SAMToGBSdbPlugin reads a SAM file to determine the potential positions of Tags against the reference genome. The plugin updates the current database with information on tag cut positions.
3.5 DiscoverySNPCallerPluginV2
DiscoverySNPCallerPluginV2 takes a GBSv2 database file as input and identifies SNPs from the aligned tags. Tags positioned at the same physical location are aligned against one another, SNPs are called from the aligned tags, and the SNP position and allele data are written to the database.
3.6 SNPQualityProfilerPlugin
This plugin scores all discovered SNPs for various coverage, depth and genotypic statistics for a given set of taxa.
3.7 ProductionSNPCallerPluginV2
This plugin converts data from fastq and keyfile to genotypes, then adds these to a genotype file in VCF or HDF5 format. VCF is the default output.
4 Variant Detection
4.1 Variant Call Format (VCF)
A variant call format (VCF) file is the most common way to communicate a list of variations from the reference genome. VCF is a flexible text based format that is often stored in a gzip compressed format (*.vcf.gz extension). A detailed description of the nuances of this file format can be found at here. Briefly, the format contains meta-information lines, a header line, and then data lines. Each data line is tab-delimited with 8 fixed fields per record, described in the table below. The INFO field is the fixed field that contains a large amount of metadata about the variant. The data contained in the INFO field is variable and will be specified in the VCF header section.
Following the 8 fixed fields, there are optional fields describing the genotype information for each sample (the same types of genotype data must be present for all samples). A FORMAT field is given specifying the order of the colon-separated genotype data for each sample field. See the VCF documentation for more information about genotype fields.
| Col | Field | Description |
|---|---|---|
| 1 | CHROM | Identifies the chromosome on the reference |
| 2 | POS | The reference position, with the 1st base having position 1. |
| 3 | ID | Semi-colon separated list of unique identifiers where available |
| 4 | REF | Reference base(s) |
| 5 | ALT | Comma separated list of alternate non-reference alleles |
| 6 | QUAL | Phred-scaled quality score for the assertion made in ALT |
| 7 | FILTER | A semicolon-separated list of codes for failed filters (PASS or . indicate that the variant passed quality checks) |
| 8 | INFO | Additional information encoded as a semicolon-separated series of short keys with optional values |
| 9 | FORMAT | A colon-separated field that specifies the data types and order for the following <SAMPLE> fields |
| 10+ | <SAMPLE> | The genotype information for the sample specifed in the header line |
The table below provides the number of SNPs that were found per contig:
| Chromosome/Contig | Number of SNPs |
|---|---|
| 1 | 22115 |
| 2 | 21824 |
| 3 | 40542 |
| 4 | 39876 |
| 5 | 21850 |
| 6 | 19540 |
| Total SNPs: | 165747 |
Interpretation: The table above indicates the number of SNPs discovered per Chromosome/Contig. These results indicate the unfiltered SNP marker sites detected by the Tassel v2 GBS pipeline . To generate high quality filtered SNPs you will need to perform traditional site and sample filtering. Depending on your scientific question and organism of interest you may consider following the GATK best practices for variant filtering. A few commonly used variant filtering programs you might consider using are the GUI based program Tassel or the command line programs bcftools, GATK, and vcftools.
5 Sample Relationships
5.1 Principal Component Analysis
Above we used the PCA method to visualize the genetic distance and relatedness of the organisms in the population you provided for GBS. To perform an accurate PCA analysis it is necessary to filter SNP sites, cull weak samples and and impute any missing data. We pre-filter you SNP data for: SNP sites with genotype information at >=60% of the samples in your population. Next, we cull any samples containing inadequate genotype information (<=80% of the pre-filtered SNP sites). Finally, we select only SNP sites containing sufficient genotype data in the population for imputation (i.e. sites with data for >=80% of samples). We then perform whole genome phasing and impute any missing data using the extensively tested Beagle algorithm on these high confidence SNP marker sites before generation of the PCA. During de novo SNP discovery, SNP phasing and imputation are impossible due to the lack of contiguous stretches of variants. Therefore, in de novo analyses, we accept only SNP sites which have data in the entire culled sample population. Information on the number of SNPs and filtering steps used to generate this plot can be found in the ./hapMap/PCA_analysis/Filter.log. Above we have utilized only the first three principal components (which often explain much of a GBS populations genetic variance) for visualization. If you would like further principal component data on your samples, principal components 1-5 calculated by the sklearn PCA function are present in the ./hapMap/PCA_analysis/PCA1.txt file.
Interpreting your PCA:Each point on the PCA represents an individual in your GBS population. If you hover over a point with the mouse, the chart will display the corresponding sample name and principal components (PC1,PC2,PC3 are x, y, and z respectively). Additionally, you can drag the 3D plot to better visualize relationships of the different clusters. Pictures from any angle can be saved by clicking the appropriate options in the upper right corner. Each point is colored based on the unsupervised clustering of the first three principal components. We have optimized the agglomerative clustering of the PCA components in your data by testing the clustering with up to 10 data clusters and picking the optimal number clusters such that: if another cluster was added, less than 10% of the samples would be present within. Although we have optimized the PCA to show the largest differences in the population occasionally the automated clustering method doesn't recognize patterns that are visible upon closer inspection. Additionally, if all samples in your population are very closely related, the clustering (defined by color) may have little meaning. The interpretation of the relationships of samples will require viewing the members of each cluster and their relationship in distance to nearby individuals within and outside their cluster. Finally, one of the strongest measures of the accuracy of the PCA is likely your own knowledge of the relationships of the organisms being sequenced. Using known outgroups within your GBS population will provide a reasonable measure of how much distance on the PCA is equivalent to a sizable genetic distance.
How does PCA work?The PCA method is performed via eigenvalue decomposition of the data covariance or correlation matrix after a data normalization step. Effectively, PCA is a rotation of the axes of the original coordinate system to a new orthongonal axes (principal axes), such that the new axes coincide with the directions of maximum variation for the original observations. More generally, PCA is a mathmatical transformation on a large number of correlated variables into a small number of uncorrelated variables (principal components), i.e. a process of dimensionality reduction. The first principal component accounts for the largest amount of variation possible, the second accounts for less and so on and so forth until all the variance within the dataset is explained. For further information on PCA see the PCA wikipedia page.
5.2 Principal Component Variance
![[PCA variance]](data:image/png;base64,iVBORw0KGgoAAAANSUhEUgAAB4AAAAWgCAYAAAC/kV7ZAAAABHNCSVQICAgIfAhkiAAAAAlwSFlzAAAuIwAALiMBeKU/dgAAIABJREFUeJzs3Xn8VFX9P/A3m4DsIKgssoiCGyK4oRSguKEmKqmpCSqCYmJqapkgqGWgFZVLpoKYGcUiKUYuICAuJIsIuGKYigiCgOzbZ35/8GO+n/nsK+jt+Xw8eDzmzpx75szcO/dzua97zqmQSqVSAQAAAAAAAMC3XsU93QAAAAAAAAAAyoYAGAAAAAAAACAhBMAAAAAAAAAACSEABgAAAAAAAEgIATAAAAAAAABAQgiAAQAAAAAAABJCAAwAAAAAAACQEAJgAAAAAAAAgIQQAAMAAAAAAAAkhAAYAAAAAAAAICEEwAAAAAAAAAAJIQAGAAAAAAAASAgBMAAAAAAAAEBCCIABAAAAAAAAEkIADAAAAAAAAJAQAmAAAAAAAACAhBAAAwAAAAAAACSEABgAAAAAAAAgIQTAAAAAAAAAAAkhAAYAAAAAAABICAEwAAAAAAAAQEIIgAEAAAAAAAASQgAMAAAAAAAAkBACYAAAAAAAAICEEAADAAAAAAAAJIQAGAAAAAAAACAhBMAAAAAAAAAACSEABgAAAAAAAEgIATAAAAAAAABAQgiAAQAAAAAAABJCAAwAAAAAAACQEAJgAAAAAAAAgIQQAAMAAAAAAAAkhAAYAAAAAAAAICEEwAAAAAAAAAAJIQAGAAAAAAAASAgBMAAAAAAAAEBCCIABAAAAAAAAEkIADAAAAAAAAJAQAmAAAAAAAACAhBAAAwAAAAAAACSEABgAAAAAAAAgIQTAAAAAAAAAAAkhAAYAAAAAAABICAEwAAAAAAAAQEIIgAEAAAAAAAASQgAMAAAAAAAAkBACYAAAAAAAAICEEAADAAAAAAAAJIQAGAAAAAAAACAhBMAAAAAAAAAACSEABgAAAAAAAEgIATAAAAAAAABAQgiAAQAAAAAAABJCAAwAAAAAAACQEAJgAAAAAAAAgIQQAAMAAAAAAAAkhAAYAAAAAAAAICEEwAAAAAAAAAAJIQAGAAAAAAAASAgBMAAAAAAAAEBCCIABAAAAAAAAEkIADAAAAAAAAJAQAmAAAAAAAACAhBAAAwAAAAAAACSEABgAAAAAAAAgIQTAAAAAAAAAAAkhAAYAAAAAAABICAEwAAAAAAAAQEIIgAEAAAAAAAASQgAMAAAAAAAAkBACYAAAAAAAAICEEAADAAAAAAAAJIQAGAAAAAAAACAhBMAAAAAAAAAACSEABgAAAAAAAEgIATAAAAAAAABAQgiAAQAAAAAAABJCAAwAAAAAAACQEAJgAAAAAAAAgIQQAAMAAAAAAAAkhAAYAAAAAAAAICEEwAAAAAAAAAAJIQAGAAAAAAAASAgBMAAAAAAAAEBCCIABAAAAAAAAEkIADAAAAAAAAJAQAmAAAAAAAACAhBAAAwAAAAAAACSEABgAAAAAAAAgIQTAAAAAAAAAAAkhAAYAAAAAAABICAEwAAAAAAAAQEIIgAEAAAAAAAASQgAMAAAAAAAAkBACYAAAAAAAAICEEAADAAAAAAAAJIQAGAAAAAAAACAhBMAAAAAAAAAACSEABgAAAAAAAEgIATAAAAAAAABAQgiAAQAAAAAAABJCAAwAAAAAAACQEAJgAAAAAAAAgIQQAAMAAAAAAAAkhAAYAAAAAAAAICEEwAAAAAAAAAAJIQAGAAAAAAAASAgBMAAAAAAAAEBCCIABAAAAAAAAEkIADAAAAAAAAJAQAmAAAAAAAACAhBAAAwAAAAAAACSEABgAAAAAAAAgIQTAAAAAAAAAAAkhAAYAAAAAAABICAEwAAAAAAAAQEIIgAEAAAAAAAASQgAMAAAAAAAAkBACYAAAAAAAAICEEAADAAAAAAAAJIQAGAAAAAAAACAhBMAAAAAAAAAACSEABgAAAAAAAEgIATAAAAAAAABAQgiAAQAAAAAAABJCAAwAAAAAAACQEAJgAAAAAAAAgIQQAAMAAAAAAAAkhAAYAAAAAAAAICEEwAAAAAAAAAAJIQAGAAAAAAAASAgBMAAAAAAAAEBCCIABAAAAAAAAEkIADAAAAAAAAJAQAmAAAAAAAACAhBAAAwAAAAAAACSEABgAAAAAAAAgIQTAAAAAAAAAAAkhAAYAAAAAAABICAEwAAAAAAAAQEIIgAEAAAAAAAASQgAMAAAAAAAAkBACYAAAAAAAAICEEAADAAAAAAAAJIQAGAAAAAAAACAhBMAAAAAAAAAACSEABgAAAAAAAEgIATAAAAAAAABAQgiAAQAAAAAAABJCAAwAAAAAAACQEAJgAAAAAAAAgIQQAAMAAAAAAAAkhAAYAAAAAAAAICEEwAAAAAAAAAAJIQAGAAAAAAAASAgBMAAAAAAAAEBCCIABAAAAAAAAEkIADAAAAAAAAJAQAmAAAAAAAACAhBAAAwAAAAAAACSEABgAAAAAAAAgIQTAAAAAAAAAAAkhAAYAAAAAAABICAEwAAAAAAAAQEIIgAEAAAAAAAASQgAMAAAAAAAAkBACYAAAAAAAAICEEAADAAAAAAAAJIQAGAAAAAAAACAhKu/pBgDFt2bNmpg+fXp6uVmzZlG1atU92CIAAAAAANjztmzZEp9++ml6uUuXLlG3bt092CLY/QTA8C00ffr06Nmz555uBgAAAAAAfKNNnDgxzjnnnD3dDNitDAENAAAAAAAAkBACYAAAAAAAAICEMAQ0fAs1a9YsY3nixInRunXrPdQaAAAAAAD4Zli8eHHGFIo5r6fD/wIBMHwLVa1aNWO5devWcdhhh+2h1gAAAAAAwDdTzuvp8L/AENAAAAAAAAAACSEABgAAAAAAAEgIATAAAAAAAABAQgiAAQAAAAAAABJCAAwAAAAAAACQEAJgAAAAAAAAgIQQAAMAAAAAAAAkhAAYAAAAAAAAICEEwAAAAAAAAAAJIQAGAAAAAAAASAgBMAAAAAAAAEBCCIABAAAAAAAAEkIADAAAAAAAAJAQAmAAAAAAAACAhBAAAwAAAAAAACSEABgAAAAAAAAgIQTAAAAAAAAAAAkhAAYAAAAAAABICAEwAAAAAAAAQEIIgAEAAAAAAAASQgAMAAAAAAAAkBACYAAAAAAAAICEEAADAAAAAAAAJIQAGAAAAAAAACAhBMAAAAAAAAAACSEABgAAAAAAAEgIATAAAAAAAABAQgiAAQAAAAAAABJCAAwAAAAAAACQEAJgAAAAAAAAgIQQAAMAAAAAAAAkhAAYAAAAAAAAICEEwAAAAAAAAAAJIQAGAAAAAAAASAgBMAAAAAAAAEBCCIABAAAAAAAAEkIADAAAAAAAAJAQAmAAAAAAAACAhBAAAwAAAAAAACSEABgAAAAAAAAgIQTAAAAAAAAAAAkhAAYAAAAAAABICAEwAAAAAAAAQEIIgAEAAAAAAAASQgAMAAAAAAAAkBCV93QDAAAAgP8NH3/8cbRs2TK9/PLLL0fXrl33XIPYbbp27RrTp0+PiIjevXvH448/vmcblEB9+vSJ0aNHR0REly5dYtq0aXmWe/zxx+Pyyy9PL6dSqd3RvD2qvPY/+3X5SqVSsWDBgli8eHGsXLkyVq1aFXvttVfUq1cvDjjggOjYsWPUq1dvTzfzW2/Hjh2xePHiWLx4cXz22Wexdu3a2Lp1a9SuXTsaNWoU7du3j4MPPjgqVtSXbJcPPvggFixYEF988UV8/fXX0bBhw9h3333jwAMPjLZt2+6W72rdunUxc+bM+O9//xurV6+OunXrRpMmTaJz585Rv379EtWZSqVi9uzZ8eabb8batWtjn332iRNPPDEOPfTQYte1dOnSeOSRRyIiokWLFtGnT58StQkoOQEw8K2WlZUVq1at2tPN+NZp0KCBE3cAyCbnxfCcKlasGFWrVo2aNWtGo0aNokWLFtG2bds4/vjjo1u3btGgQYPd2NrCTZw4Md56662IcMGlJN56662YOHFiennIkCF7rjEAsIdlv7kgPzVr1ow6derEwQcfHMccc0xcdNFFcdRRR5Xo/WbNmhV/+MMf4sUXX4wVK1YUWPawww6LSy65JH74wx9G06ZNS/R+u8u2bdti4cKFMXv27PS/BQsWxLZt29JllixZEi1atNgt7fnZz34WM2bMiLlz58bmzZsLLNu0adO44oor4ic/+UnUqlVrt7SvLNx8881x3333ZTw3atSoEp0bb9u2Le6///4YOXJkLFy4MN9ydevWjdNPPz1GjhwZ1atXL/b7FGbRokUxZMiQmDhxYmzfvj3X61WqVInTTjsthg0bVqzgdu7cuXHllVem/w+R3cknnxyPPvposfbNgQMHxoQJEyIiYtKkSUVeDyg7AmDgW23VqlXRqFGjPd2Mb50VK1ZEw4YN93QzAOBbIysrKzZt2hSbNm2KL7/8MhYtWhTPPfdcRERUrlw5TjnllLj++uvjtNNO28Mt3WnixIkZvcAEwMXz1ltvxdChQ9PLAmAAKNj69etj/fr1sXTp0nj55Zdj+PDh0b1793jkkUeKHBp99NFH8eMf/7hYYdGiRYvitttuiyFDhsSAAQNi0KBBJe79WJ6OO+64mD9/fmzZsmVPNyXt3nvvjR07dhSp7GeffRZ33nlnPPbYYzFu3Lg4/vjjy7l1pTd37tz47W9/WyZ1zZkzJ3r37h2LFi0qtOyaNWtizJgx8Yc//KHMA+CHH344rrvuuoybBnLatm1bTJo0KV588cUYMWJEXH311YXWO3v27DjppJNi3bp1eb4+ZcqUOOGEE+L111+P5s2bF1rfv/71r3T427NnzzjzzDMLXQcoewJgAACAHBo1apSrd8PGjRtjzZo1sWnTpoznt2/fHpMnT47JkyfHySefHCNHjowDDjhgdzYXAGC3ad68eVSunHlZed26dfHll19mDCn+0ksvxdFHHx0zZ86Mtm3bFljnjBkz4rzzzss1ytsBBxwQp5xySrRq1SoaNmwY27Zti+XLl8cHH3wQL7zwQnz11VcREbF169YYMWJENG/ePH784x+X0SctO//+97/3dBPyVbVq1Tj66KPjiCOOiAMPPDDq1q0blSpVipUrV8a8efNi0qRJ6WBw6dKlccopp8TMmTPjyCOP3MMtz9/27dvjyiuvLHLAXZDXX389Tj/99Pj666/TzzVr1izOPPPMOOigg6J+/fqxbt26+OCDD+KNN96IOXPmlMvQ+g8//HCuMLdTp07Ro0ePaNy4caxduzZmzJgRzz77bOzYsSO2bNkSAwYMiDp16sQPfvCDfOvdvn17XHLJJeltvP/++0e/fv3igAMOiHfffTcefvjhWLduXSxbtiyuuOKKmDJlSoHt3Lx5c/zoRz+KiIgaNWrE7373u1J+cqCkBMAAAAA5DBs2LN9esxs3bow5c+bErFmz4oknnogFCxakX5syZUp06NAhpk+fHocddthuai18e7Ro0eJ/Yr5R2BMef/xxc9CyW0ybNi3PXr2rVq2KcePGxaBBg+LLL79MP9ezZ89YuHBhrtB4lylTpsQZZ5yR0avx+OOPj2HDhsV3v/vdfNuxY8eOmDJlSgwZMiRef/310n2o3ahGjRrRvn37OProo2Px4sXpUWV2t+uuuy7OPPPM+O53vxt77bVXvuXWrFkT1113XTz55JMRsbO391VXXfWNDrXvu+++9FDGhxxySLz77rslqmfx4sVx2mmnpcPR2rVrx7333ht9+/bNd2q1ZcuWxciRIwv8Totr0aJFMXDgwPRy1apVY9SoUbmC3RtuuCHmzZsXZ599dixdujRSqVRcfvnl0alTp3x74k+cODE++OCDiIho3LhxzJ07N/bdd9/065dffnl07NgxNm/eHFOnTo3Zs2fH0UcfnW9b77nnnvjoo48iImLw4MFujIU9yASQAAAAxbD33nvHd77znfjJT34Sb7/9djz//PPRrFmz9OurVq2KHj16xLJly/ZgKwEAdq8GDRpE//79480334z99tsv/fz777+fDg9z+vzzz+Piiy/OCH9vvPHGeO211woMfyMiKlWqFKeeemq89tprMXbs2KhTp07ZfJBycN1118Xjjz8eCxcujK+//jpmzpwZI0aMKDBIK2+//e1vo3v37oUGlXXr1o0nnngiTj755PRzb775ZsyfP7+8m1giH374YXoqj4YNG8bdd99donpSqVRceeWV6fC3Vq1a8cILL0S/fv3yDX8jdvag/fnPfx61a9cu0fvm5c4774ytW7emlx944IF8e/UeddRR8c9//jO9Xbds2RI///nP8637n//8Z/rxrbfemhH+RkQceuih0a9fv/Ty5MmT861r8eLFMWzYsPR6N9xwQwGfCihvegADibP/T9+JijX32dPN+MbIWr8ylv3q0D3dDABIrFNPPTXeeuutOP300+PNN9+MiIhPPvkkrrzyyowLKgXZsWNHLFq0KN59991YtmxZbNiwIWrWrBn77LNPdOzYsdBhE8vLmjVr4u23344PP/wwvvrqq9i2bVvUq1cvmjZtGieeeGKp5th79913Y968ebF8+fLYsGFDVK1aNerUqRPNmzePI444Iho3blyiehctWhTz58+PFStWxJYtW2LfffeNww8/PDp27BgVKlQocXu/zXbn/rVhw4aYMWNGfPbZZ/Hll19GnTp14oILLoiGDRuWqt6tW7fGggUL4r333ovly5fHpk2bonbt2tGoUaM49thjo2XLlmX0CSK++uqrmD59enz66aexefPmaNSoUXTu3Dlat25dqnrXr18fM2fOjM8++yxWrlwZFStWjAYNGkTbtm2jY8eOUa1atWLXuWTJknjzzTfTv6OGDRtGmzZtolOnTlGpUqVStbc4Fi1aFLNnz45ly5ZF3bp1o1mzZtG1a9eoUaNGmb5HWf62N27cGHPnzo133nkn1qxZE9u2bYu99947GjVqFK1atYr27duXaO7G7du3x6xZs2Lx4sWxcuXK2Lp1a9SpUydat24dHTt2jAYNGhS7zl0WLVoUCxYsiGXLlsWWLVuiQ4cOceqpp5a4vrxkZWXFzJkzY/HixbFixYqoX79+HHLIIXHiiScWGHgURXkdn3fH/lcSX331Vfp4uHHjxmjcuHF07ty5yPPhfps1b9487rnnnrj88svTz/3jH//Ic3SVfv36xYoVK9LL1157bfz6178u9nv26tUr2rdvn+55WJDyOgcpyO9///syr3N3qlChQgwcODBj+N9///vf37hhoFOpVFx11VWxefPmiNgZcpf0fPXJJ5+MGTNmpJeHDRsWxx13XJm0szjWr18fTz/9dHr5kEMOiSuvvLLAddq1axe9e/eORx55JCIi/vrXv8bw4cOjSZMmucouXLgw/fi0007Ls77TTz89vQ9nL5/Tj370o/Q81w8++GBUqVKlwHYC5UsADCROxZr7RKWapbvABABQHPXr148JEyZEhw4d0kMeTp48Od544404/vjj81xn3bp1MWHChBg/fnxMnz49Y16xnJo1axY33nhjDBgwIN9eGh9//HGeIdj06dPzvbDeu3fvXMOFvv/++zFmzJh49tlnY968eZGVlZXnuhUqVIiTTjopBg8eXGgPnV1SqVQ88sgjcd9998WHH35YYNkWLVpEz5494ze/+U2hwcCWLVvi/vvvj9///vfxySef5FmmSZMm8bOf/SyuvvrqfIOxFi1axH//+99cz+f3/s2bN4+PP/64wLblZcWKFdGkSZPYvn17REQMHTo0Bg8eXOT1V65cGY0bN073lvr5z3+eq3dLWe9f2XXt2jWmT58eEf+3D61atSpuvvnm+Pvf/x4bNmzIKH/YYYdF165dIyL3fvryyy+nX8vrc/7973+Pp59+Ol599dVc829n17Zt27j11lujd+/eRQqSsm/rO+64I4YMGRIrV66MG264IcaOHZu+eJld586d44EHHoh27doVWn92r776atx1110xderUjB5u2VWrVi1OPvnkGDhwYKGBXlZWVowePTqGDx8e7733Xp5lGjRoEAMHDoxbbrmlRMFyUb366qtx3XXXxbx583K9VrNmzejdu3cMHz489t577xLVX1a/7exWrFgRgwYNiqeeeirWr1+fb7nKlSvH8ccfHzfeeGOce+65hdb78ccfx1133RXjxo3L9/dWsWLF6NixY/Tp0ycGDBiQ6/Vp06ZFt27d0stLliyJFi1axPjx42Po0KEZUw5ERHTp0iVjf+nTp0+MHj06/dq0adMKbXd2f/jDH2LYsGGxdOnSXK81btw47rzzzkJDh5zKYxvuUt77X0mtXLkyrr/++hg/fnyex5KuXbvGAw88EIcemveN2mvXro3GjRvHxo0bIyLilltuSfeoK4q1a9fG/vvvnz5mFnf9snLeeedF375903Owzp07N1eZ+fPnZwx/fPDBB8e9995b4vds3bp1vjfrlNc5yP+Sgw8+OGN51znvN8kjjzySPkfp3r17XHLJJcU+Fu5y//33px+3bt06+vfvXxZNLLaZM2dmnD+cf/75RVrvggsuSAfAqVQqJkyYENddd12ucqtXr04/zm+45ubNm+dZPruxY8fG888/HxERl112WXTp0qVI7QTKjyGgAQAAykDTpk3j1ltvzXhu+PDh+ZYfP3589OnTJ5599tkCw7mIiE8//TRuuOGG6NatW7lfbOvfv38MGTIk5syZk2/4G7HzQtKUKVOia9euRRpab+vWrdGzZ8/o379/oRdeI3aGKSNGjEhfOM7Phx9+GEcccUT85Cc/yTdciIhYunRp/OhHP4qTTjqp0O+7vDVq1Cijh0V+w2LmZ8yYMRkXAn/4wx/mKrM796/58+dHu3btYtSoUbnC39K4//7749prr42XXnqpwPA3IuK9996Lyy+/PM4999wStWH+/PnRvn37ePLJJ/MMbCJ2XoDt3LlzvPHGG0Wqc+vWrXHllVdG586d4/nnn883/I2I2Lx5czz33HPxy1/+ssA6ly9fHp06dYorrrgi3/A3YudQ9HfccUd07NgxPv/88yK1t7juv//++O53v5tn+Baxs8fSAw88EMcdd1x88cUXxa6/PH7bCxYsiMMPPzz+9Kc/FRj+RuzsyTtz5swYP358oW198MEHo02bNjFy5MgC25CVlRVvvvlmXHvttYXWuct1110XvXr1yhX+lqWsrKy44IILYuDAgXmGvxE7h+nt27dvXHzxxQX+bciuPI/P5b3/ldS7774bHTp0iKeeeirfY8m0adOiQ4cOMXHixDxf3zVywi6jR49O3zBUFH/5y18yjpl9+/Yt8rplqXbt2rHPPv83Oltef1/uu+++jOWf/exnJep5X5jyOgf5X7NrKORdChvRYMiQIVGhQoX0v/Keo/zzzz+PW265JSJ23lj1xz/+scR1vf322xlzHF955ZWlHgWhpJYsWZKxXNQb0XKWe+aZZ/Isl/1GsfyOwWvXrs2z/C7r169PD/dct27dUt3IAZQdPYABAADKyFVXXRV33HFHOoCaMmVK7Nixo9AeTfXr14/OnTvHUUcdFY0aNYrq1avHqlWrYvbs2fHMM8+kL+S+9tprccEFF8SUKVNyXYSqUqVKHHjggRGxs4fbrot01apVy3O4t4jINcdXToccckh06tQpDjnkkKhXr15s3749Pv3005gyZUo6BEulUjFo0KBo0qRJxlCPOd1xxx0ZF54aNGgQZ599dhxxxBFRv3792LZtW6xatSoWLVoUM2fOLFLP2kWLFkXXrl1j5cqV6eeaNWsW55xzTrRt2zaqVasWS5YsiXHjxsX7778fEREzZsyIM844I6ZPnx6VK2f+l7hFixZRuXLlWLduXcZwlLu+15yaNm1aaBvzc9lll6V7PX344YcF9hbP6c9//nP68bHHHhtt2rQpsHxZ7F/5Wb16dZx77rnx+eefR+XKlaNHjx7RuXPnaNCgQaxcuTKmTZtWJkMR77///nHiiSdG+/btY5999okqVarE8uXL4/XXX4/Jkyenw5F//OMfMWDAgHQvyKJYvnx5nH322bF06dKoWbNmnHPOOXHMMcdErVq14tNPP42//e1v8e6770bEzovfF198cSxcuLDAXoXbt2+PHj16ZAyVWaFChTjxxBPjpJNOSv8mly9fHnPmzImpU6fmurCe07Jly6Jz587xn//8J/1cw4YN43vf+160a9cuatasGZ999lk888wzMWfOnIiIeOedd6JLly4xZ86cMp2LcMyYMTFw4MBIpVLp50444YQ488wzY999940VK1bE5MmT45VXXomFCxfGJZdcUuTQMKLsf9sRO0P2c845JyOEOvLII+PUU0+NVq1aRfXq1WP9+vWxdOnSmD9/fkyfPr1INxPccccdceedd2Y816ZNm+jRo0e0atUq9t577/jqq69i/vz5MW3atPjss8+K/D2MGDEi3QOtVatW0bNnz2jdunVUqFAhFi9eXGbh/tChQ2Ps2LERsbOHX69evaJly5axbt26mDFjRkyaNCn9G/vrX/8aderUiYceeqjAOstjG+5S3vtfSW3cuDF69eoVn376aVSsWDFOO+20OPnkk6Nu3brxySefxIQJE9JDp27ZsiUuvPDCeOmll+I73/lOrrr69++fDsuWL18ekyZNip49exapHY8++mj6cZcuXeKggw4q/YcroezBdc6/BalUKmOqjDp16sSFF15YLu0oj3OQ/0U5pzbp3LnzHmpJ3q699tp0UDlo0KB8z9+K4oUXXshY7tGjR6naVho5e9zWq1evSOvlLPfWW2/lWa5x48bpY9P777+f5/8Pdh2nd5XPafDgwekbiH75y19Go0aNitRGoJylgG+dhQsXpiIi/W/hwoV7ukl7zIoVKzK+i4hINbl7ReqAESn//v+/Jnfn/o5WrFixpzcdAHyjjBo1KuNv5ahRo0pcV/fu3TPq+ve//53ve3bp0iU1adKk1NatW/Otb8WKFamzzz47o86RI0cW2IbevXuny3bp0qVY7T/jjDNS119/feqdd94psNzUqVNT++/oBtsBAAAgAElEQVS/f/p9atWqlVqzZk2eZTds2JCqXr16uuzZZ5+dWrduXYH1z5kzJ3XZZZeltm/fnufrGzduTB122GHpOitWrJi655578vwut2/fnho6dGjGd3jnnXfm+94594fysGnTplSdOnXS7zFgwIAirff+++9ntO3+++/Ps1x57l9dunTJdX7Zpk2b1KJFiwpt/5IlSzLWe/nll/MtO3To0NQ555yTevnll1M7duwosM5OnTpl1Dt16tQC29G8efOMfSciUt27d08tW7YsV9lt27alrrjiioz6H3jggQLrv+mmmzLKH3rooalZs2blW37jxo2p0aNHp/r165fn6zt27EidfPLJGXVef/31qfXr1+dZ/k9/+lOqUqVK6bJXXHFFge0tjuXLl6fq16+frrtq1aqpMWPG5Fl2/PjxqWrVqmV8zxGR6t27d771l9dvO/vvukKFCoXu55s2bUr99a9/TQ0fPjzfMs8++2yqQoUK6Xpr166deuqpp/Itn5WVlZoyZUrqpJNOyvP1l19+OddvKyJSQ4cOTW3btq3A9qZSRT/25zzG7do2t912W57H3NmzZ6eaNm2asc5LL72Ub/3leXwu7/2vuLIfD3e9xz777JOaMWNGrrJZWVmp4cOHZ3zWgw46KLVp06Y8627Xrl263FlnnVWk9sybNy+j/j//+c+l+ny7ZN+3IiK1ZMmSQtf58ssvM34frVq1ynh9/vz5GXWeccYZZdLWnMrjHKQs3XHHHcX+bveE1157LVWjRo10O08//fRC18n52Upzfl2YsWPHpt/nsMMOyzje5Dy2FqUd3//+9zPOcXedh8yZMyd17bXXpg499NBUrVq1UjVq1Ei1aNEi1bNnz9TDDz+c2rhxY5l/tt/85jcZ7X/mmWeKtN7atWtz/T3J63pg9u3Uq1evPOs66qij0mUef/zxjNfefvvtVOXKlVMRkTrmmGMKPGfbnVw/h1TKENAAAABl6IQTTshYzmvOu4iIiy66KKZNmxZnnnlmVKlSJd/6GjZsGOPHj48TTzwx/dwf/vCHsmlsHiZMmBAjRoyIQw45pMBy3bp1i8mTJ6fbvm7dunx7Xb7xxhvpXqaVK1eOkSNHRs2aNQusv0OHDjF69Oh8e4/ee++9sWjRovTyH//4x/jpT3+a53dZqVKlGDx4cNx0003p54YNG5YxnN3uVq1atfj+97+fXv7b3/5W4BDBu2Tv/VulSpV8e0vtzv2rXr16MXXq1HznsyypW265JSZOnBhdu3YtsEdyixYt4l//+lfGvI/F+QxZWVnRoUOHeO6552K//fbL9XrlypXjwQcfjBYtWqSfe+qpp/Ktb9GiRfGb3/wmvXzooYfGK6+8Escee2y+61SvXj0uu+yyePjhh/N8/cknn8zoTXzbbbfFiBEjokaNGnmWv+qqq2LEiBHp5VGjRhVp2NOiuOeee+Krr75KLz/88MP57ofnnXdejBo1KiKiyD0wy+u3PXXq1PTjc845p8ARCyJ2/kYvuuiiuPnmm/N8fdu2bXHNNdeke6FWr149Xn755fjBD36Qb5275k7Pvi0Lc+utt8bgwYML7BFbWllZWXH11VfHL37xizyPuR07dozJkydH1apV08/l971ElO/xubz3v9LIysqKihUrxj/+8Y88e/VWqFAhbr755vj5z3+efu7DDz+MP/3pT3nW169fv/TjyZMnF6nH9675PiN2Hpt79epVnI9Qpp588smMXtrHHXdcxuvZh9eNiAKPkaVRHucg/wt27NgRq1atimnTpsWAAQOiS5cu6VERWrVqFY899tgebuH/WbNmTXpu2woVKsTDDz9c4LlPUWQfXv6ggw6KzZs3x7XXXhtHH310PPDAA/HOO+/EunXrYsOGDfHxxx/HxIkTo3///tGqVauYMGFCqd47p4YNG2Ys5xwSOj/ZRwzZ5aOPPsr13KWXXpqe63rcuHHxi1/8IjZv3hwRO4d+vvrqq9PfR+3ateO8885Lr5tKpeKaa66J7du3R8WKFeOhhx7aY0NlA7n5NQIAAJSh7CFRRGQMf5ldXvNn5adKlSpx1113pZfnzZsXy5cvL1H7ClOcdh155JEZYcfkyZPzLJd9/sX69etnzAlYElu2bIkHHnggvXzqqafGVVddVeh6d911V3q+ug0bNmSEqXvCZZddln68atWqXEMr5pRKpeIvf/lLerlHjx75fpe7c/8aNGhQnsMBllZxPkPt2rXjtttuSy+/8MILxZq78f7774+99tor39erVq0affr0SS/PmTMn3zk5f/3rX6dDjwoVKsTo0aOjfv36RW5LfnXucsghh2Rsr/xce+21cdhhh0XEzn0nv3C5OLZs2RJPPPFEerlTp07Ru3fvAte56KKLolu3bkWuv7x+29mPQwcffHCR2lOQv/71rxnDOd99993RoUOHUteb3b777htDhgwp0zrzUr9+/fjVr35VYJnDDz88Bg4cmF6eN29eeqjx7MpzG5b3/lcW+vTpk+tGsJxuv/32aNasWXo5e2ib3aWXXpoean7Hjh2Fzp+6adOmjJtTLrnkkmIdR8vSnDlzYvDgwRnP5Qyjc/6dadmyZbm0pazPQZJq5cqVGfP1Vq5cOfbZZ5/o1q1bPPTQQ7Ft27aoVKlSXHrppTFr1qxy+btfUjfddFN6O1911VUZN7WVVPbpAho1ahTf//7348EHH0z/fd9rr72iadOmUbdu3Yz1vvjii+jVq1f87ne/K3Ubdjn66KMzll966aUirZdXubzm+G3dunX86Ec/Si/ffvvtUb9+/WjevHk0bNgw4/zh7rvvjlq1aqWXR40aFa+++mpERFxzzTXRsWPHIrUN2D0EwAAAAGUo53xb2XsqlcZ3vvOdjN5Xb775ZpnUW1rdu3dPP86vTdnnSl2xYkWevQ+K48UXX8yYo/eGG24o0nrVq1ePCy64IKOePalz584ZF7wLC6RnzpyZ0esje4BcWiXdvypWrFim7SiN7Pvihg0b0vP2Fubggw+OTp06FVou+xzNmzdvznOOyKysrIyePyeffHKuC7fFtXDhwnj77bfTywMHDixS75oKFSpkbJuy2N9fffXVjGNaUYK9iMyejAUpz9929uPQ66+/XqR6C/L3v/89/bhOnTpx9dVXl7rOnC666KLdEuBdeOGFUadOnULL5dyO2edU3aU8t2F5739loX///oWWqVatWsZvc+HChXn26Ms5J+7IkSMzetTmNG7cuFizZk16uajfT1lZv359zJ49O2699dbo3Llzxrzmxx57bJx77rkZ5VetWpWxnDNIKytlfQ7yv6pBgwbx0EMPxeOPP17kEH3IkCGRSqXS/7LfSFVWpk6dGiNHjoyInTfNDBs2rNR1plKpjP13ypQp6Zv02rRpE08//XR8/fXX8emnn8bq1avjvffeyxhVIpVKxY033lis0R4K0rZt24wbTP/5z3/GO++8U+A6GzduzLgZZ5fsnyu7++67L+MmjU2bNsUnn3ySMTrNT37yk3RP64id/8e59dZbI2Lnd/+LX/wiV70rV66MadOmxaRJk2L27Nn53jwHlA8BMAAAQBnKOaxgXnfal8Sunhi7LF26tEzqLa39998//XjVqlWxZcuWXGWOOuqojOVzzjmnVAH2K6+8kn5crVq1OOmkk4q8bvYhJmfNmlXiNpSFChUqxKWXXppenjRpUsbF+5yyB8T16tWLs846q8zaUtL9q02bNulee3ta9n0xouifoSjhb0REkyZNMpbz2lbz58/PGLr2/PPPL1LdBcm+v0dEnHnmmUVeN/v+vmjRovTwnSWVc8jW0047rUjrnX766UUqV56/7ey9c1955ZUYMGBArgCqqLKystI9niJ2fr7sIVNZKawnaVkp6nZs3bp1HHjggenlvI7j5bkNy3v/K6169eoVeRjjnG3K729i9vD6o48+imnTpuVbZ/YheY855pho165dkdpSEi1btszoLVqhQoWoVatWHHPMMTF8+PD08LERO0dGmTBhQnqI2V1yBlGFDctcUmV9DpJUlSpVigMPPDD9r1mzZhnHtVWrVkW/fv2iTZs2e/wGul02bdqU8RsZMWJEmdxIsGHDhoxh43eFoB06dIhZs2ZFz549M26aa9OmTYwcOTKGDx+efi4rKyt+/OMfF3jTRnFkv5lmx44dcdFFF2X0Us5u+/btceWVV+Z5o9qu4dBz2muvvWLs2LExduzYOO2002KfffaJKlWqxP777x/nn39+TJs2Le69996MdX7605+mRzr69a9/nXEj0X/+85/o2bNn7LffftGtW7c4++yz45hjjomGDRvGL37xC0Ew7CYCYAAAgDKU84JmYb2qsrKyYurUqTFw4MD47ne/G02aNIlatWpFxYoVc11czR5oFRQUloXVq1fHo48+GhdffHG0a9cuGjZsGNWqVcvVplNOOSVjvbza1bx58zjjjDPSy4sWLYpjjz022rVrFz/72c/iX//6V7Hm450/f3768UEHHVTg0L057bvvvunHy5cvL9K8u+Upey+wLVu2ZPQqzG7Lli0xduzY9PKFF15YpM9d3vtX9nl3y8vWrVvj2Wefjf79+0enTp1iv/32ixo1auRqf875/or6GfKa9zcvOcOJvMLUnD1yStv7NyJzf69Vq1bG0LGFyb6/79ixI5YtW1aqtrz33nvpx/Xq1SvyEKB169aNpk2bFlquPH/bffr0ybhg/9BDD0WTJk3irLPOit/97ncxd+7cIg8bvmzZsoz9qyy2c152x+8rIuKII44oUdns+8Mu5bkNy3v/K63DDz+8yGVzfud5fZcRO0ceyB7kPvroo3mWW7x4ccyYMSO9vLt7/+alSpUqcfnll8fcuXNz3UATERnDyEbs7EFcHsr6HCSp6tWrF4sXL07/++STT2L9+vXx7rvvxuDBg9N/Az/66KM4/fTTY/To0Xu4xRGDBw9O9+g+/fTT46KLLiqTeqtXr57ruYoVK8aTTz5Z4Hn9zTffnHHTy8KFC4s8XHNh+vfvH8ccc0x6ecGCBXHUUUfFQw89lO6pu3Llynj66afjhBNOiDFjxkRE7nOXnL+7nHr16hX/+te/4ssvv4ytW7fG559/HuPGjYsuXbpklHvjjTfSx6Nu3brFJZdckn5t4cKFcfzxx8c//vGPXH9X16xZE7fffnucf/75xZqqAyiZynu6AQAAAEmSM3QqaO7PGTNmxDXXXFPoMG55yd67pizt2LEjfvWrX8Uvf/nL2LhxY7HXz69djzzySHTt2jUWL16cfm7BggWxYMGC+NWvfhUVK1aMjh07xplnnhk//OEPo1WrVvm+R/ZeewsWLMjVq6g41qxZEw0bNizx+qXVunXr6NSpU3pI2j//+c95Dlf67LPPZuxbRRl2eXfsX7Vr1y523cXx9NNPx/XXXx+ffvppsdct6mco6RC7efXqydmjNGev5JLIXue6detKtb+vXr26VG3Jvn6jRo2KtW7Dhg0z5szNS3n+tg844IB47LHHok+fPumeR1u2bInnnnsunnvuuYjYecNOly5d4vvf/36cd955+fbqLY/tnJfy/n3tUpxtmf07zWt/Ks9tWN77X2kVp0116tSJvfbaK7Zu3RoRBf82+/Xrl56fc8KECbF69epc0008+uij6WNSjRo1yiwIy0/z5s2jcuX/u6xcoUKF2HvvvaNOnTpx0EEHxTHHHBPnnntuRqifU87RI8rzxrayPAf5JpkwYULccssthZYbOHBgxhzeRVWhQoVo27ZtDB06NPr06RPdu3eP//znP5GVlZUOJA899NCSNL3U5syZE7/97W8jYucw3w8++GCZ1V2pUqWoVq1axnnEqaeeGoccckih6/74xz+OqVOnppdffPHFXDdLlkTVqlVj/Pjx0b179/jggw8iYudIJwMGDMh3nTZt2sTFF18cd9xxR/q5nMeOktixY0dcc801kUqlokqVKhlDTW/dujUuvPDCdO/kvn37xm233Rb7779/zJo1K/r37x/vv/9+PPPMM3Hfffelh5AGyocewAAAAGXoP//5T8ZyfvOkTZw4Mbp3755nOLf33nvH/vvvHy1btswYji/7xdayGlIuu1QqFZdcckncfvvteYa/9evXj6ZNm2a0KWcPrPza1aRJk5g9e3bcdNNNefY+yMrKijfffDOGDBkSBx98cPTt2zff4bPL8iJxSULuspY9zH311VfznAsy+/DPu0Ljguyu/Sv7OmXtgQceiPPOOy/P8LdWrVrRuHHjaNWqVcZnyK48fiOFKY8hTb9J+3v2Xs959ZAqSI0aNQotU96f9ZJLLonXX389unfvnmcwuXbt2njmmWfSAVB+Pdx219C15fn7yq442zL7dsyrx2Z5bsPy3v9Kq7htyn6DQUG9Xy+99NJ02c2bN8df/vKXjNe3b9+esa9edNFFhfbyK61p06Zl9Bb98MMPY/78+TFjxox47LHH4uqrry4w/I2IXK/nNVxtWSnLc5Bvkq+//jo++uijQv9lnzu7pFq2bBnjxo1Lz0G/ZcuWuOeee0pdb0ls3749+vbtm+5Bescdd0TLli3L9D1y3oDTrVu3Iq3XpUuXjL8vc+fOLbM2NWvWLGbNmhWXXXZZVKpUqcCyvXr1ildeeSXXzXBFnb+5IPfff3+89dZbEbFzXuDswfjYsWPT555nnHFGPPLII9GyZcuoVq1adOnSJZ5//vn0iC3Dhg3Ld0hqoGzoAQwAAFCGdvXk3CWvoUG//PLLuOKKK9LDW1auXDn69u0bvXr1ig4dOuR7d37z5s3jk08+KftG/3+PPvpo/O1vf8t4v4EDB0b37t2jTZs2GcOn7vLyyy8XeY7HOnXqxH333RdDhw6NF154IaZOnRqvvvpqzJ8/P2OutR07dsRjjz0W//73v2PmzJm5LsJlv2hes2bNQi8yF2R3BSwFufDCC+P666+PrVu3RiqViieffDIGDRqUfn3VqlUxefLk9PIPf/jDAuv7pu5fxfHuu+9mzHdXs2bNGDBgQJx11lnRvn37PC/gp1Kp9IXpPSVnELh+/fpCh4EvTPb9vVq1ankOpVpUJe3tvEv2EK24F22LMv/w7vhtH3300fHiiy/GkiVLYvLkyTF9+vSYOXNmfP755xnlli9fHn369IklS5bEkCFDMl7Lazt/m23atKnIIXb27ZjXOuW5Dct7/yut4rYpe8Bd0Pdfp06duPDCC2PUqFERsXOu3109giMinnvuufjiiy/Sy3379i1WO/aUnPMl55zjuayV1TnI/7KjjjoqvvOd78T06dMjYue+l0qlStXTvyQefPDBdAB5xBFHxI033ljm73HggQfGihUr0ssHHHBAkdarXbt21KtXLx265zdPb0nVrVs3Ro8eHYMGDYoJEybEzJkzY9myZbFx48bYb7/94sgjj4yLL744/f+P7MPL77333tGmTZtSvf+yZcti8ODBEbHzvPH222/PeH38+PHpx3n1Tm/evHn84Ac/iCeeeCJWr14dU6ZMibPOOqtUbQLyt+f/pwsAAJAQa9eujTfeeCO9XLdu3Wjfvn2uciNHjkwP91ixYsWYNGlSnHbaaYXWX97z/v7mN79JPz700EPjtddeKzS8KkmbatSoEeeee26ce+656TpeeumlGDNmTEycODHdo2PBggVx1113xb333puxfvbeCyeccEI8//zzxW7DN0m9evXirLPOigkTJkTEzt6+2QPgMWPGpMPcChUqFBoAf1P3r+L4/e9/n/7M1atXj5kzZ8aRRx5Z4DrfhPbnHNJ02bJlpQpsIzL39wMOOCDef//9UtVXGtlvHsh+YbwoinIRfHf+tlu2bBkDBgxID5/5wQcfxKRJk2LkyJGxaNGidLk777wzzj///Iw5W/Pazt9mK1asKHIAnH075nUzSXluw/Le/0qrOG1au3ZtevjniMKHZe3Xr186AH7rrbdizpw50bFjx4jInBf48MMPj+OPP744zd5jjjjiiKhfv346KHvttddi8+bNpb5RpTClPQf5JunTp0/06dNnt75n+/bt0wHw6tWr46uvvsp1TCxv2UfbWbp0abRt27bA8jlvzrj11lvj7rvvTi//8Y9/jO7du2eUOeywwzJu6izOfpm9bHlN2dK6deu45ZZbCh0CPPv/Szp06FBoz+HC3Hjjjene8b///e9zTZUwZ86ciNh5vprfaDUnnHBCPPHEE+nyAmAoP4aABgAAKCN/+tOfMnoZde/ePc8eiVOmTEk/PuWUU4oUzq1cubJchyP8/PPPM3oJDBo0qEg9F3MOeV0SdevWjV69esW4cePi1VdfzRhG88knn8xVPvuFvpLMb/tNlH0Y6A8//DBmzZqVXs4+/HPnzp0LHebwm7h/FVf2z3DZZZcVGv5GlM2+WFqHH354xvLs2bNLXWf2/X3JkiV7dNjy7D2HVq9enavXbH7WrFlTpPlX9+Rv++CDD44bb7wxFixYEDfddFP6+VQqFU899VRG2SZNmmQEdmWxnfekBQsWFLnswoUL04/z6klWntuwvPe/0sr+3RS3bGG98o4//vho165devmxxx6LiJ1/u7OPEPFt6f0bsTMg6tGjR3p5zZo18fe//323t6O45yD/63KOBrMrMN9Tvvrqq0KHwM55rFixYkXG63mN4pDzBs6iDqOdSqUy5vTe3eF4dvPmzcu4QSlnyF1cU6ZMiTFjxkRExNlnnx3f+973cpXZdbPNPvvsk+fIQRE7h7Lepbg38wDFIwAGAAAoA5999lkMHz4847n87spfunRp+nFRgq2IiKlTpxa5Lbvm1oqIjGENC5K9TcVpV/agriwcd9xx0a9fv/TyF198kXEhLSJzHrbPPvss5s2bV6ZtyP79RRT9OyyNHj16ZPSc2xX65gyDswfF+Snv/Wt3KMlnKOt98f+xd+fxMZ77/8ffk0SCINaEEFJL7Tu1VW1V1NZSqpZaauspammr7bFVlarWTqkiR784BMdWy0FViZ1aYqnaYwtijURIMr8//MzJnXWSzJgYr+fjkUdd19zXdX9mMpOk857rutOiQoUKypkzp6UddyvEtIr7fH/8+LFDV7zXqFHD0P7vf/9r1Thra7b3a9saJpNJ48ePN7xpHz/INJlMevXVVy3tDRs2ZIjriaeVtd+fM2fO6PTp05Z29erVExxjz++hvZ9/6XX79m2rtzHesGGDoZ3YYxlf3N+NixYtUmRkpAICAiwBnIeHR4o7RGQ0n3zyiaH97bff2m3FpDWs+RvkRXfu3DnLv11dXW1yTdmMqGXLlob20y2nU/LXX38ZVhwXLVrUpnWlRtzdAVxdXfXBBx+kea5Hjx7po48+kvRkK+mpU6cmetzTn0fJbYkf9/eloz9AADg7AmAAAAAASKdbt26pTZs2unnzpqWvRYsWSb6hazabLf+29o3OKVOmWF1P3K087969a9WYuDVZW9fJkyft8sZ6/JVQT7cCfqpJkyaGkO3bb7+16fnjb4Vq7WOYHpkyZdK7775raT/d9jnu6t/MmTOrXbt2Kc5l7+fXs5Da+/Do0SP9+OOP9izJKi4uLobv0ZYtW9K9OrRKlSoqUaKEpT1+/Ph0zZcederUUe7cuS3tOXPmWDXO2uPs/dq2lqurq4oVK2Zpx/8ZJEkdOnSw/Pvu3buaNWvWM6nNHpYuXWrVDgA//fSTod26desEx9jze2jv558tWHOuqKgoy/an0pOdA6wJiTp37mzZbvXu3bsKDAzUvHnzLLe3adPG8Pg8DypWrKjmzZtb2idOnNDnn3+e5vlOnz6d7r9LUvob5EV2//59wwcvKleunOhON/Y2efJkmc1mq7+2bt1qGD9//nzD7W+99VaCcxQuXNhwneoVK1YoOjo6xdqWLFliaDds2DCN9zJ9Tp48aQiAW7durUKFCqV5vu+++85yCYphw4bJ398/0eOefngqPDw8yVXTFy9eTHA8APsgAAYAAACAdNi0aZMqVaqkffv2Wfr8/f0Nb7rEV6RIEcu/169fn+Kn3ydPnqydO3daXVPcN2X+/vtvRUVFpTimcOHChvbatWuTPT4qKkrdunWzanVsSEhIqj7hf/jwYcu/PT09lS9fPsPt2bNnV//+/S3tpUuXGt4Et1ZSwWL8N7VSs61nesRd3RsWFqZ169YZtp9s1aqVVdty2/v59SzEvQ8pPRelJ6vtz58/b8eKrDd48GCZTCZJT4Lsrl27Wr11ZGJcXFwMgciePXs0cuTIVM9ji1V1Hh4ehufpzp07U9wiNTAw0OrV2fZ8bafm+REZGalTp05Z2om90d2+fXvDz83hw4fr4MGDqaozowgLC9OXX36Z7DHHjx83rPiqXLmy5Rq0cdnze2jv558tzJ8/33DNzcSMHTvWEID06tXLqrm9vLwMHxT67LPPdObMmVTPk9H89NNP8vb2trSnTJmizz77LMEH01KybNkyVatWTSdOnDD02/pvEGeR2LbHKfn4448NH4p75513kj1+1KhRMplMlq+AgIBUn9OR4q5Qv3z5siZOnJjs8SEhIZo0aZKlnSNHDsu1phNTv359w+Njq79jbt++rffee89ynXFPT0/98MMPaZ7v3LlzGjt2rKQn2/zHvUxCfHG3qk9qS/e4/dbu8gIgbQiAAQAAACAVIiIitGPHDn3//feqUKGC3njjDYWEhFhuz5cvn9atWycfH58k54h7Tda///5b/fv3T3RVwePHj/XNN99o8ODBkmT1KouaNWta/h0ZGakvv/wy2a3YJCl//vyGN2HGjRunTZs2JXrsxYsX1aRJE+3Zs8eqmubOnauSJUtqypQpunbtWrLHLl261HJtQ+lJ6Pk0TItr6NChhuut9uzZU59//nmKq9hu3bql+fPnq1q1alq5cmWix5QpU0bZs2e3tEeOHKnQ0NBk57WFV155xbDy6JNPPjFstWjN9s+S/Z9fz0Lc+/Dbb79Z3niM78GDB+rfv7+mTCHW/esAACAASURBVJmSYeovVaqUhg4damkfP35cdevWNXxIJL6HDx/q//7v/9S3b99Eb+/atavh2n2jR49Wjx49Urx2Xnh4uAIDA9WwYUObrfL+4osvDNe/7dmzpwIDAxM9duXKleratask659f9nptN2zYUC1bttTatWuTXdEXERGhHj166M6dO5a+xFaHubm5afbs2ZafTxEREWrYsKHl+oiJMZvN+v3339W4ceNk78uz5uLiohkzZmjEiBGJBmV//vmnmjVrZghlJ0yYkOR89vz5bO/nX3q4uLgoJiZGrVq1UlBQUILbzWazJk6cqK+//trSV7x4ccOWwymJe2zc30vFihVT/fr101a4g/n6+mrRokWGyy9MmDBBdevW1Y4dO5IdGxMTo02bNqlOnTpq165dojt22ONvEGfQuHFjffzxxzpy5EiKx4aEhKht27aaP3++pc/Pz0/9+vWzZ4kO165dO9WqVcvS/vLLLzV79uxEjz19+rQaN25seA4OGTLE8PMqvWbMmJHiB/Z2796t1157zbBl9YQJE5JcsWuNAQMGWP4/YubMmXJ3d0/y2BYtWlj+PX78eIWFhRluX7ZsmeXSJpkzZ1ajRo3SXBeAlLk5ugAAAAAAyGiGDh2qMWPGGPoiIyN1586dZK/z2LhxY82dO1d+fn7Jzv/BBx9o3LhxlvDmxx9/1ObNm9WuXTsVK1ZMUVFROnXqlP7zn//owoULkp686btx40ZLOzk1atRQ2bJldezYMUnSxIkTNWPGDPn7+ytz5syW41q1aqXRo0db2l9++aVldVFkZKSaNGmiZs2aqWHDhsqTJ49u376tXbt2ac2aNXr48KHc3Nw0fPhwq1YjnjlzRgMHDtTgwYNVrVo1vfLKKypatKhy5cqlx48f6/z58/rvf/9r2C7X09Mzwfch7m3/+c9/9Nprr+nq1asym80aP368Zs6cqSZNmqhatWqW69LduXNHZ86c0aFDh7Rv374Ut/DLlCmTunTpopkzZ0qStm7dKl9fXxUpUkTZs2e3vBns6+urdevWpXjfU6NLly4aNmyYJBmutent7W0IRZNj7+fXszBo0CDNnj3bEjj985//VGBgoN5++235+fkpIiJCwcHBWrFiheV+jhkzJsVVjM/K119/rQMHDlg+RHH8+HHVqFFDderUUaNGjeTr6ysXFxeFhobqzz//1ObNm3X37l3Vq1cv0flcXV3173//W3Xr1rWsbps/f74WLVqkxo0bq0aNGvLx8ZGbm5vu3r2r8+fP69ChQ9qzZ4/lMXzjjTdsct+8vb01c+ZMdezYUWazWVFRUWrfvr3q1Kmj5s2by8fHR9evX9eGDRu0bds2SU/C18ePH2v79u0pzm+v13ZsbKzWrl2rtWvXKmfOnKpdu7aqVq0qHx8fZcuWTffu3VNwcLBWrlxpCNabNWuW5GPXtGlTjRo1yvIz8O7du3rvvfc0atQoNW/eXEWLFlXWrFl169YtBQcH67fffjOs/Mwohg0bptGjR+vrr7/W0qVL1a5dO/n7++v+/fvavn271qxZYwjN+/btm+yb9vb8+Wzv5196tG3bVsHBwTpx4oRee+01y+9PLy8vXbp0ScuXL9fRo0ctx7u7u2vu3LmG38spqVmzpipUqJAgtOvZs+dzHVQ2atRImzZtUtu2bS2BUVBQkOrWrasiRYqocePGKlq0qPLly6fHjx8rNDRUp06d0saNG63aYcHWf4OkxdSpUxO9bmr8+uvXry83t4Rv2S9cuDDBdbDTIzIy0lJT8eLFVb16dZUtW1a5c+dW1qxZ9eDBA124cEF79+7VH3/8YdjxJUeOHAoMDJSnp6fN6smoFi1apFq1aunatWuKiYlR3759NXv2bLVu3Vp+fn4KDw/X7t27tWLFCsOOO40aNdI///lPm9ayZs0a9evXTwULFlSDBg1Uvnx55c2bV1FRUbp8+bI2bdqkffv2GVbPjxo1Sh9++GGaz7ly5UrLTiydOnUyXOc9Me+//77GjBmj0NBQnT9/XpUrV1afPn2UP39+7dmzx7AjRK9evdgCGrAzAmAAAAAAiOf69esprqx7ys3NTY0bN9bAgQOtDliyZ8+uZcuWqWnTppZA+e+//05ylWOrVq00bdo0vfzyy9bdAUm//PKLmjZtarkfUVFRlmt3PVWpUiVDu3379tqxY4emTZsm6clqpXXr1iUacnp4eGj27NmGrXqtERsbq71792rv3r3JHpc7d26tWrUq2esiFi9eXPv371ebNm0sqwnu37+vZcuWadmyZSnW4urqmuRtY8eO1Z49e3TgwAFL3XFX40oyrBC0lS5dumj48OEJtr587733En1DODHP4vllb0WKFFFAQIA6d+5sCYQOHTpkWNESV9++ffXFF19kmADYzc1Na9euVe/evfWvf/1L0pPX044dO1Jc0ZaUPHnyaPfu3erUqZPlzdioqChLqJmS5J7vqdWhQwfduHFDAwcOtIQCQUFBia56LFu2rBYuXGi4Zm5K7Pnalp68dpP62RZXo0aNElzPMb4RI0Yod+7cGjx4sCUk/euvvxL8vM3IRo4cqRMnTigwMFB//fVXsqFXhw4dNH369BTntOf30N7Pv7TKmjWrli1bpiZNmujSpUv69ddf9euvvyZ6rLu7u5YsWaLXXnst1efp3bu3YeWlm5ubunXrltayM4x69eppz549+vjjjw2P24ULF5K9rMZTmTNn1scff6zu3bsneYwt/wZJrVu3bhm27E5KUh/ESmknl/Q4ffq04UNnySlfvrz+9a9/qXLlynarJyPx9/fXhg0b9NZbb1m2aP7zzz/1559/JjmmTZs2WrBggU1/78Z1+fLlFLe/z5Ytm8aNG5euVdoREREaOHCgpCdb0H///fcpjsmRI4cWLFigli1b6tGjRwoJCbF8sDGuihUraty4cWmuDYB1Msb+SAAAAACQgZlMJnl4eChv3rwqU6aMmjVrpiFDhigwMFChoaFat25dqlfX1a1bV3v37k32k/TFihXTjz/+qFWrViW73VpiKleurODgYI0bN04NGjRQ/vz5rVplNHXqVM2ZM0cFCxZM9HZXV1e9/vrr2rt3r2VrzZR06dJFI0aMUPXq1VMMMXPlyqV+/frp5MmTevXVV1Oc29fXVzt37tS///1vVa9ePcUVUEWLFtWHH36o7du3q127dkke5+XlpZ07d2r+/Plq1aqV/P395enpafcVVoULF050Fai12z8/Ze/n17Pw7rvvauvWrYleZ/SpcuXKKTAwUD/++OMzrMw67u7uCggI0NatW1W/fv1k3wjOmjWr3n77bY0aNSrZOXPkyKE1a9Zow4YNatCgQYqvJ19fX3Xt2lXr16+3bPVtK/3799e2bdsSfJDkqWzZsumjjz7S3r17lT9//lTPb+vX9syZM9WjR48E1ztPTPny5fXzzz9r06ZNhu3gk9KvXz+dOHFCnTp1UtasWZM8ztXVVXXq1NGcOXNSnPNZcnFx0dKlSzVlypQkf/b7+vpqzpw5Wrx4sdWhhr1+Pkv2f/6lVZkyZfTnn3+qY8eOSf5crV+/vg4ePJjo1uLW6Ny5s+F70KJFi2d6H+2pWLFiWrt2rXbv3q2OHTsarg2cGJPJpAoVKui7777TuXPn9O2338rLy8twjD3/BnmejRkzRu+//36Ku9ZITx7nOnXqaO7cuTp48OALE/4+VbFiRR09elRDhgyx7F6QmHLlymnx4sVatmyZXVZHt2rVSlWqVEl2S/vcuXOrZ8+eOnHiRLq36B49erTlAwljxoyx+ufMG2+8oY0bN6pEiRKJ3v7077sXYQU54Ggmc/yPFQPI8I4dO2a4nk5wcLDKli3rwIoc58aNGwn+h6DgmOtyzZbPQRVlPDHhN3R5mPExun79uvLl4zECACCjOHPmjIKCgnT16lW5urqqQIECKlWqVLLBl709fvxYe/bs0ZEjR3T79m3lzJlTBQoUUO3atdP1RnNkZKQOHz6sM2fOKDQ0VBEREcqcObPy5MmjcuXKqWLFiukKI2/evKmgoCBdu3ZNt27dkqurq7y8vPTSSy+pbNmySYYbziwjPr9S69ixY9qzZ4+uX78uDw8PFShQQBUqVFCZMmUcXZrVbt26pR07dujy5cu6deuW3N3dlS9fPpUsWVJVqlSRh4dHque8d++egoKCdOnSJcu2qTly5FCRIkVUpkwZvfTSS7a+G4kKDg7Wvn37FBoaKi8vL/n5+alBgwY2fXPXlq/tK1euKDg4WOfPn9ft27f1+PFjZc+eXQULFlSVKlXSteovKipKQUFBOnfunG7cuCFJypkzp4oXL66qVava9HqQ9hAbG6vt27fr9OnTun79unLnzq1SpUqpbt266b6Orr1+Pj+L519a3Lp1S3/88YdCQkIUEREhX19fvfrqq+l+Xe7evdtwXdK1a9eqefPm6S03QzKbzTpy5IhOnz6tmzdvKiwsTO7u7sqVK5f8/f1VtWpV5cyZ0+r57P03yPPq2rVrOnbsmM6fP69bt24pKipKnp6e8vLyUvHixVWpUiXlyJHD0WVmCNHR0QoKCtLZs2cVGhoqDw8P+fj4qFatWs/sd+7t27e1f/9+Xbt2TdevX1dsbKzy588vf39/1apVy+odY5ITHR2t7777To8ePVLmzJn12Wefpfp3QExMjHbu3KlDhw4pPDxcPj4+atiwYbquR5wavH8OEAADzyV+gf0PAXDKCIABAAAAAHAOvXr1smyJXKhQIZ0/f95uW80CwPOK988BtoAGAAAAAAAAgAzvzp07Wrx4saXds2dPwl8AAJAoAmAAAAAAAAAAyOB++OEHPXjwQJKUKVMm9erVy8EVAQCAjIoAGAAAAAAAAAAysEWLFum7776ztHv06CFfX18HVgQAADKy9F8RHAAAAAAAAABgM6tXr9aIESMUGxurkJAQ3blzx3Jbrly5NGrUKMcVBwAAMjwCYAAAAAAAAADIQG7duqXDhw8n6Hd3d9fChQuVP39+B1QFAACeF2wBDQAAAAAAAAAZlKurq3x9fdWpUyft379fzZo1c3RJAAAgg2MFMAAAAAAAAABkIN26dVO3bt0cXQYAAHhOsQIYAAAAAAAAAAAAAJwEATAAAAAAAAAAAAAAOAm2gHZSly5dUnBwsC5evKjbt29LknLlyiUfHx9Vr15dvr6+djlveHi4du3apVOnTunOnTvKlCmTfHx8VKFCBVWqVEkmk8ku502rq1evas+ePTp37pwePHigLFmyqHDhwqpWrZpeeuklR5cHAAAAAAAAAAAApAoBsJMIDQ3V6tWrtWXLFm3dulXXr19P9viiRYuqR48e6tOnj/LmzZvu8x89elRjxozRypUr9ejRo0SP8fX1VZ8+fTRkyBB5enqm+5zpsWbNGo0fP147d+6U2WxO9JiKFStq8ODB6tKlS4YLrgEAAAAAAAAAAIDEsAX0c27//v1q2LChfH191bt3by1ZsiTF8FeSzp49q2HDhqlo0aKaN29ems9vNps1ZswYValSRUuXLk0y/JWkK1euaOTIkSpTpoz27t2b5nOmR3h4uN555x21atVKQUFBSYa/knT48GF17dpVDRo0UGho6DOsEgAAAAAAAAAAAEgbAuDn3P79+7V161bFxsamafz9+/f1wQcf6MMPP0zT+H79+mn48OGKjo62eszFixdVv359/fHHH2k6Z1qFh4fr9ddf1/Lly1M1btu2bXr11Vd17do1O1UGAAAAAAAAAAAA2AYBsBMqXry4PvzwQy1ZskRHjhzRtWvXdOfOHZ08eVLz5s1TzZo1E4yZNWuWRo4cmarzzJgxQzNnzjT0ZcuWTUOGDNHevXt18+ZNnTt3TqtXr1bjxo0Nx0VGRqpNmzYKCQlJ/R1Mox49emjPnj2GvsKFC2vy5Mk6evSobt++rePHj2vOnDkqXbq04bjTp0+rbdu2qQq6AQAAAAAAAAAAgGeNawA7CTc3N3Xo0EE9e/ZUvXr1Ej3Gy8tLJUuWVPfu3TV79mz1799fjx8/ttw+duxYtW/fXmXLlk3xfKGhofrss88MfQULFtTGjRsN4/PkySN/f3+1bNlSP/zwgz799FPLtsthYWEaNGiQli1blpa7nCpr1qxRYGCgoa9evXpatWqVvLy8LH05c+ZU6dKl1aVLF3Xp0sUwZufOnZo1a5b69etn93oBAAAAAAAAAACAtGAF8HPOxcVF7733nk6cOKFffvklyfA3vj59+mj27NmGvujoaI0ePdqq8d98840iIiIsbVdXV61evTrZ8HjIkCEaMGCAoW/FihU6ePCgVedMK7PZrOHDhxv6ChcurLVr1xrC37g8PDy0ePFiVa1a1dAf/34DAAAAAAAAAAAAGQkB8HPugw8+0KJFi1S8ePFUj+3evXuCwHj9+vV69OhRsuNCQ0MThMf9+vVTlSpVUjznN998I19fX0vbbDbr66+/TkXVqbdq1SodPnzY0DdlyhRly5Yt2XGurq6aM2eOoe/atWsJ+gAAAAAAAAAAAICMggD4Oefq6pqu8V27djW079+/ryNHjiQ7ZtWqVYaQ2GQyqX///ladz9PTUz169DD0rV+/XuHh4VZWnHpLly41tP39/dWqVSurxlauXFmvvvqqoS/+VtIAAAAAAAAAAABARkEA/IKrWLFigr6rV68mO2bVqlWG9quvvqpixYpZfc5u3boZ2lFRUdq4caPV41Pj8ePHWr9+vaGvS5cucnGx/qkfv95du3bp+vXrtigPAAAAAAAAAAAAsCkC4BdclixZEvQ9ePAgyeMfPXqkLVu2GPrq16+fqnMWK1ZMhQoVMvT9+uuvqZrDWrt27dKdO3cMfamtN/7xsbGx2rBhQzorAwAAAAAAAAAAAGyPAPgFd/HixQR93t7eSR5/6tQpRUVFGfrq1KmT6vPGH3P06NFUz2GN+NtZu7q6qkaNGqmao1ixYvLx8TH02ateAAAAAAAAAAAAID0IgF9wv//+e4K+5LZzPnnyZIK+0qVLp/q88cf89ddfqZ7DGvHrLVy4sDw9PVM9T/x6E3scAAAAAAAAAAAAAEcjAH6BxcTEaOHChYa+0qVLq0iRIkmOOXHihKHt6uqqggULpvrcfn5+hvb9+/d1+fLlVM+Tkvj1Fi5cOE3zxK+XABgAAAAAAAAAAAAZEQHwC+znn39WSEiIoe/dd99NdsyVK1cM7QIFCsjV1TXV504siLVHABy/3vhBrrXi12uPWgEAAAAAAAAAAID0IgB+QV26dElDhw419OXKlUv9+/dPdlx4eLihnSNHjjSdP7Fx8ee2BXvVGxkZqdjY2DTXBQAAAAAAAAAAANiDm6MLwLMXHR2tjh076u7du4b+sWPHKnfu3MmOjR+oZsmSJU01JDbuWQTAtq43rYFyXNevX9eNGzdSNeb06dPpPi8AAAAAAAAAAACcDwHwC2jgwIHavn27oa9Jkybq06dPimMfPHhgaGfOnDlNNTyrANje9doiAJ45c6a++uqrdM8DAAAAAAAAAAAAsAX0C2batGmaMWOGoa9QoUL65ZdfZDKZUhxvNpsNbWvGWDNPeuZKjeetXgAAAAAAAAAAACA1CIBfIEuWLNHAgQMNfV5eXvr111+VL18+q+bIli2boR0ZGZmmWh4+fJigz9PTM01zJSf+nBm9XgAAAAAAAAAAACA92AL6BbFhwwZ16dJFsbGxlr4sWbJozZo1qlChgtXz2CoATmxc/LltIVu2bLpz506y57WGPev9xz/+oXbt2qVqzOnTp/XWW2/Z5PwAAAAAAAAAAABwHgTAL4AdO3aobdu2evz4saUvU6ZMCgwMVN26dVM1V/zQ8969e2mqKbFx9gqAUzqvNeKPy5Ili1xcbLOA3tvbW97e3jaZCwAAAAAAAAAAAC82toB2cgcPHlSLFi0UERFh6XNxcdGCBQvUvHnzVM/n6+traF+9elUxMTGpnickJCTFuW0h/pyJndca8cfZo1YAAAAAAAAAAAAgvQiAndixY8f0xhtv6O7du4b+WbNmqUOHDmmas1SpUoZ2TEyMLl++nOp5Ll68aGhnz55dhQoVSlNNyYlfb/zzWiv+uNKlS6e5JgAAAAAAAAAAAMBeCICd1JkzZ9S4cWOFhYUZ+r///nv16tUrzfMmFnyeOHEi1fOcPHnS0C5ZsmSaa0pO/HovXryoBw8epHqe+PXGD5YBAAAAAAAAAACAjIAA2AmFhISoUaNGunr1qqF/1KhRGjJkSLrmfvnll+Xh4WHo27lzZ6rnCQoKMrTLly+frrqSUqFCBUM7JiZGe/fuTdUcZ86c0bVr1wx99qoXAAAAAAAAAAAASA8CYCcTGhqqRo0a6cKFC4b+wYMHa+TIkeme393dXY0aNTL0/f7776ma4+zZswmuqZuW6xFbo1atWsqZM6ehL7X1btu2zdB2cXFR06ZN01saAAAAAAAAAAAAYHMEwE7k9u3baty4sf7++29Df58+ffTDDz/Y7DytW7c2tLdv366zZ89aPf5f//qXoe3h4aEmTZrYpLb4MmXKpGbNmhn6fvnlF5nNZqvniF9vrVq15O3tbZP6AAAAAAAAAAAAAFsiAHYS4eHhatq0qY4ePWro79y5s2bOnGnTc7Vu3Vru7u6Wttls1vTp060aGxERoblz5xr6mjVrpmzZstm0xrjat29vaJ87d05r1qyxauzhw4f1xx9/GPratWtns9oAAAAAAAAAAAAAWyIAdgIPHz5Uy5YtE1zbtk2bNgoICJCLi22/zT4+Purdu7ehb9q0afrzzz9THDts2DBdvnzZ0jaZTBo+fLhV561fv75MJpPhyxqtW7dWxYoVDX0DBgxQeHh4suNiYmLUq1cvQ1/+/PkT9AEAAAAAAAAAAAAZBQHwcy46Olrt2rVLcF3bZs2aafHixXJ1dbXLeYcNG6asWbMa6mjdurWOHz+e5JhJkyZp8uTJhr42bdqoSpUqdqnxKZPJpK+//trQd+HCBbVq1Ur37t1LdMyjR4/UuXNn7du3z9D/z3/+03C/AQAAAAAAAAAAgIzEzdEFIH1GjhyptWvXGvrc3NxUokQJjRgxIk1zVq1aNcVtjn18fDR+/Hj179/f0hcSEqKaNWuqb9++evfdd/XSSy8pPDxcR44c0bRp0/Tf//7XMEfu3Lk1ceLENNWYWi1btlTbtm21fPlyS9/WrVtVoUIFDRkyRI0aNZKvr69CQ0MVFBSkH374IUGYXatWLfXt2/eZ1AsAAAAAAAAAAACkBQHwcy7udspPRUdHa+rUqWmes2vXrlZd57Zfv346fvy4fvzxR0vf/fv3NWHCBE2YMCHZsVmyZNGKFStUuHDhNNeZWvPnz1dISIhhq+wLFy5owIABKY4tVqyYli9fLjc3XjIAAAAAAAAAAADIuNgCGukyY8YMffXVV6naatrPz09bt25VvXr17FhZQtmzZ9fmzZv19ttvp2pc3bp1tWPHDhUoUMBOlQEAAAAAAAAAAAC2QQCMdDGZTBoxYoQOHjyodu3ayd3dPcljCxQooFGjRun48eOqUaPGM6zyf7Jnz64VK1Zo1apVql27drLHVqhQQQEBAdq2bZvy58//jCoEAAAAAAAAAAAA0o79bJ9zAQEBCggIcHQZqlChgpYuXar79+9r586d+vvvv3X37l25urrKx8dHFStWVOXKlWUymdJ8jt9//91m9bZq1UqtWrXSlStXtHv3bp0/f14PHjxQlixZ5Ofnp+rVq6to0aI2Ox8AAAAAAAAAAADwLBAAw6ayZ8+uJk2aqEmTJo4uxSq+vr5q06aNo8sAAAAAAAAAAAAAbIItoAEAAAAAAAAAAADASRAAAwAAAAAAAAAAAICTIAAGAAAAAAAAAAAAACdBAAwAAAAAAAAAAAAAToIAGAAAAAAAAAAAAACcBAEwAAAAAAAAAAAAADgJAmAAAAAAAAAAAAAAcBIEwAAAAAAAAAAAAADgJAiAAQAAAAAAAAAAAMBJEAADAAAAAAAAAAAAgJMgAAYAAAAAAAAAAAAAJ0EADAAAAAAAAAAAAABOggAYAAAAAAAAAAAAAJwEATAAAAAAAAAAAAAAOAkCYAAAAAAAAAAAAABwEgTAAAAAAAAAAAAAAOAkCIABAAAAAAAAAAAAwEkQAAMAAAAAAAAAAACAkyAABgAAAAAAAAAAAAAnQQAMAAAAAAAAAAAAAE6CABgAAAAAAAAAAAAAnAQBMAAAAAAAAAAAAAA4CQJgAAAAAAAAAAAAAHASBMAAAAAAAAAAAAAA4CQIgAEAAAAAAAAAAADASRAAAwAAAAAAAAAAAICTIAAGAAAAAAAAAAAAACdBAAwAAAAAAAAAAAAAToIAGAAAAAAAAAAAAACcBAEwAAAAAAAAAAAAADgJAmAAAAAAAAAAAAAAcBIEwAAAAAAAAAAAAADgJAiAAQAAAAAAAAAAAMBJEAADAAAAAAAAAAAAgJMgAAYAAAAAAAAAAAAAJ0EADAAAAAAAAAAAAABOggAYAAAAAAAAAAAAAJwEATAAAAAAAAAAAAAAOAkCYAAAAAAAAAAAAABwEgTAAAAAAAAAAAAAAOAkCIABAAAAAAAAAAAAwEkQAAMAAAAAAAAAAACAkyAABgAAAAAAAAAAAAAnQQAMAAAAAAAAAAAAAE6CABgAAAAAAAAAAAAAnAQBMAAAAAAAAAAAAAA4CQJgAAAAAAAAAAAAAHASBMAAAAAAAAAAAAAA4CQIgAEAAAAAAAAAAADASRAAAwAAAAAAAAAAAICTIAAGAAAAAAAAAAAAACdBAAwAAAAAAAAAAAAAToIAGAAAAAAAAAAAAACcBAEwAAAAAAAAAAAAADgJAmAAAAAAAAAAAAAAcBIEwAAAAAAAAAAAAADgJAiAAQAAAAAAAAAAAMBJEAADAAAAAAAAAAAAgJMgAAYAAAAAAAAAAAAAJ0EADAAAAAAAAAAAAABOggAYAAAAAAAAAAAAAJwE37HH6gAAIABJREFUATAAAAAAAAAAAAAAOAkCYAAAAAAAAAAAAABwEgTAAAAAAAAAAAAAAOAkCIABAAAAAAAAAAAAwEkQAAMAAAAAAAAAAACAkyAABgAAAAAAAAAAAAAnQQAMAAAAAAAAAAAAAE6CABgAAAAAAAAAAAAAnAQBMAAAAAAAAAAAAAA4CQJgAAAAAAAAAAAAAHASBMAAAAAAAAAAAAAA4CQIgAEAAAAAAAAAAADASRAAAwAAAAAAAAAAAICTIAAGAAAAAAAAAAAAACdBAAwAAAAAAAAAAAAAToIAGAAAAAAAAAAAAACcBAEwAAAAAAAAAAAAADgJAmAAAAAAAAAAAAAAcBIEwAAAAAAAAAAAAADgJAiAAQAAAAAAAAAAAMBJEAADAAAAAAAAAAAAgJMgAAYAAAAAAAAAAAAAJ0EADAAAAAAAAAAAAABOggAYAAAAAAAAAAAAAJwEATAAAAAAAAAAAAAAOAkCYAAAAAAAAAAAAABwEgTAAAAAAAAAAAAAAOAkCIABAAAAAAAAAAAAwEkQAAMAAAAAAAAAAACAkyAAdkJhYWHauHGjxowZo1atWqlAgQIymUyGr1GjRqV5/vPnzyeYzxZfAQEBNnsM7FXzO++8Y7caAQAAAAAAAAAAgPRyc3QBsI2JEydq37592rdvn86cOePoctLE1dXV0SUAAAAAAAAAAAAAzzUCYCcxZMgQR5eQLi4uLmrQoIGjywAAAAAAAAAAAACeawTASDUvLy8NHTo0zeNv3bqlOXPmGPpef/11FSpUKL2lpUpa7kP58uXtUAkAAAAAAAAAAABgG04fAD9+/FhhYWF68OCBHjx4oOjoaGXNmlWenp7KkSOHvLy8HF2iXWTLlk1VqlRR9erVVb16dXXo0MFmc+fKlUvffvttmsdPmzYtQV+3bt3SUVHapOc+AAAAAAAAAAAAABmR0wTAjx490qFDh7R//37t379fp06d0vnz53Xt2jWZzeYkx3l6eqpIkSJ66aWXVLFiRVWrVk3VqlVTwYIFn2H16fc06H36Vbp0abm4uFhut2UAnF4BAQGGtpeXl95++23HFAMAAAAAAAAAAAA4kec6AD5z5ozWrVundevWadu2bYqKijLcnlzw+1R4eLiOHTum48eP69dff7X0Fy1aVG+++aaaNWumBg0ayMPDw+b129LevXsdXYJVgoODdfDgQUNfhw4dlDlzZgdVBAAAAAAAAAAAADiP5y4AvnnzphYtWqRffvnFECQmFvaaTCar5jSbzQnGnzlzRtOnT9f06dOVPXt2tWvXTp06dVL9+vXTVf+Lbv78+Qn6HLH9MwAAAAAAAAAAAOCMXFI+JGPYvXu33n33Xfn6+mrQoEE6cOCAJbg1m80ymUwJvqyV2FiTyWSZ+969e5o3b54aNWqkokWLatKkSbp3754d761zio6O1sKFCw19pUqVUs2aNR1UEQAAAAAAAAAAAOBcMnwAvGLFCtWsWVN16tTRsmXLFB0dbVmtm1TYGzcYTs1XfImFwefPn9cnn3wiPz8/DRo0SFeuXHkmj4MzWL9+vUJDQw19rP4FAAAAAAAAAAAAbCfDbgG9fv16DRs2TIcOHZIkQ+gbX9zwNnPmzCpVqpSKFCmiggULqlChQsqZM6eyZMmiLFmyyM3NTZGRkYqMjFRERISuXr2qS5cu6fLlyzp58qSuXbtmmPvp+eKe12w26/79+5o6dap++uknffjhh/r888+VN29emz8OziQgIMDQdnV1VZcuXRxTDAAAAAAAAAAAAOCEMlwAfPDgQX388cfauXOnpMSD37iBb5kyZVS/fn3VrVtXFSpUUMmSJeXikvaFzWFhYTpy5Ij279+vrVu3KigoSPfv3zfU8PS/ZrNZkZGRmjRpkn766ScNHTpUn376qdzd3dN8fmcVFhamtWvXGvreeOMN+fr6OqgiKSIiQrt379bJkyd18+ZNmUwm5cmTR97e3qpRo4b8/PwcVhsAAAAAAAAAAACQFhkmAL5165a+/PJLzZ07V7GxsQmC36dtDw8Pvf7662rbtq2aN2+ufPny2bSOPHnyqEGDBmrQoIE+/fRTxcTEaO/evVqxYoX+85//6OzZs5a64tYWHh6uESNGaP78+Zo8ebJatGhh07qed4sWLdKjR48Mfd27d3dQNU94eXkpOjo6ydtfeuklde7cWQMGDGB1NwAAAAAAAAAAAJ4LGeYawCVKlNCcOXMUExMjs9mc4Nq7tWvX1vz583X9+nWtWbNG3bp1s3n4mxhXV1fVqlVLEyZM0OnTp3Xw4EF99NFH8vLystQWt9azZ8+qdevWmj59ut1re57E3/45V65catWqlWOK+f+SC38l6dy5c/r6669VpEgRTZo06RlVBQAAAAAAAAAAAKRdhlkBfPv27QSrarNmzaoePXqob9++KlOmjIMrfKJSpUqaNm2avv/+ey1dulRTp07VgQMHJMkSAktPVjTjieDgYB08eNDQ17FjR3l4eDiootSJiIjQ4MGDFRQUpMWLFytTpkw2nf/69eu6ceNGqsacPn3apjUAAAAAAAAAAADAOWSYAPgps9ms3Llzq1+/furfv7/y5Mnj6JIS5eHhoS5duqhLly7atGmTxo8fr99++83RZWVI8+fPT9DXrVu3Z1+IpEyZMqlu3bp68803VblyZZUsWVI5c+ZUpkyZFBYWplOnTum3337Tzz//rCtXrhjGLl++XL179070/qTHzJkz9dVXX9l0TgAAAAAAAAAAALyYMswW0JKULVs2jRo1ShcuXNCoUaMybPgbX+PGjbV582bt2LFDderUsawCxpNtlhcuXGjoK1eunKpVq/ZM68iSJYtGjx6tkJAQbdmyRUOGDFHDhg1VsGBBeXp6yt3dXQUKFFC9evX01Vdf6dy5cxo5cqRcXIwvkYCAAM2bN++Z1g4AAAAAAAAAAABYK8MEwP369dPp06c1YsQIeXp6OrqcNKldu7a2b9+uVatWqUSJEo4uJ0NYv369QkNDDX2OWP3r4+Oj4cOHy8fHx6rj3d3dNWrUKC1YsCDBbSNHjlRUVJStSwQAAAAAAAAAAADSLcNsAT116lRHl2AzLVu2dHQJGUZAQICh7ebmps6dOzummDTo1KmTdu3apRkzZlj6Ll26pCVLluj999+3yTn+8Y9/qF27dqkac/r0ab311ls2OT8AAAAAAAAAAACcR4YJgOF8wsLCtHbtWkNfs2bNrF6Fm1GMGDFCc+fO1cOHDy19GzZssFkA7O3tLW9vb5vMBQAAAAAAAAAAgBdbhtkCGs5n0aJFevTokaHPEds/p5e3t7dee+01Q9+2bdscVA0AAAAAAAAAAACQNAJg2E387Z/z5s2rFi1aOKaYdKpVq5ahfe3aNcXGxjqoGgAAAAAAAAAAACBxBMCwi+DgYB08eNDQ17FjR7m7uzuoovSJv0VzbGyswsLCHFQNAAAAAAAAAAAAkDgCYNjF/PnzE/R1797dAZXYhtlsTtBnMpkcUAkAAAAAAAAAAACQNAJg2Fx0dLQWLlxo6KtYsaIqVarkoIrSLzQ01NB2cXFR7ty5HVQNAAAAAAAAAAAAkDg3RxeQEcTExOj27du6ffu2zGazcuXKpVy5csnNjYcnLdavX58gMH2eV/9K0q5duwxtHx8fubjw+QkAAAAAAAAAAABkLC9swrlhwwYtX75ce/fu1fHjxxUbG2u43WQyqVSpUqpRo4beeusttWjRgi1/rRQQEGBoZ8qUSZ06dXJMMTZw7do1bd++3dBXr149B1UDAAAAAAAAAAAAJO2FC4CXLFmiL774QhcuXJCU+LVdn/YfP35cJ06cUEBAgAoVKqRvvvlGnTt3fpblPnfCwsK0du1aQ1+LFi2UN29eB1WUfiNHjlRUVJShr2nTpg6qBgAAAAAAAAAAAEjaC7OHbVRUlNq2bauOHTvq/PnzMpvNMpvNMplMyX49PS4kJERdu3ZV69atFRkZ6ei7k2EtWrRIjx49MvR169bNZvP7+/sbvj/+/v4pjrl8+XKazzdv3jz99NNPhr6CBQuqffv2aZ4TAAAAAAAAAAAAsJcXIgCOiopSy5YttXLlygSh79OAN6mv+MeuXbtWLVq0SLAiFE/E3/7Z29tbb775pmOK+f8mTJigatWqafny5VZ/3yIjI/X555+rZ8+eCW776quvlCVLFluXCQAAAAAAAAAAAKTbC7EF9KhRo7R582ZLkCs92eLZzc1N9evXV7Vq1VSsWDFlz55dJpNJ9+7d05kzZ7R//35t27ZNjx8/NoTAv//+u0aMGKHx48c7+J79z4EDBxQYGGj18Zs3b9bDhw8TvS1XrlwaOnRoqmsIDg7WwYMHDX2dO3eWm5vjn2YHDhzQO++8oxw5cqhFixaqWbOmKlWqpEKFCsnLy0uZMmXSrVu3dPLkSW3dulXz5s3TjRs3EszTvXt3ffDBBw64BwAAAAAAAAAAAEDKHJ/M2dnZs2c1ceJEQ/Dr6uqqQYMGaejQocqTJ0+y42/duqXx48dr0qRJiomJsYTAkydPVp8+fVS0aNFncTdSdPTo0VQF0kFBQQoKCkr0tiJFiqQpAJ4/f36CPltu/2wL9+7d06JFi7Ro0aJUj33vvfcSbAcNAAAAAAAAAAAAZCROvwX04sWL9fjxY0lPwt9MmTJp9erV+u6771IMfyUpd+7cGj9+vFavXi1XV1dLf3R0dJpCRGcVHR2thQsXGvqqVq2q8uXLO6gi28mVK5fmz5+vRYsWZYjVzAAAAAAAAAAAAEBSnD4AXr9+vSRZruc7ZMgQNWvWLNXzNG3aVJ988ollnrhzQ9qwYYNCQ0MNfRll9W///v01ceJEtWrVSgUKFLBqTKZMmVSjRg3NmjVLly5dyjD3BQAAAAAAAAAAAEiOyWw2mx1dxFN37txRzpw5bTqnn5+frly5YgluT5w4oZdffjlNc/39998qWbKkZRtoX19fXbp0yab1wv5u3LihU6dO6eLFi7px44YePHig2NhYeXl5KVeuXPL391fVqlWVOXNmR5eapGPHjqlcuXKWdnBwsMqWLevAihznxo0b8vb2NvQVHHNdrtnyOaiijCcm/IYuDzM+RtevX1e+fDxGAAAAAAAAAJwL758DGewawCVKlNDYsWPVq1cvm81548YNQ9vf3z/NcxUpUsTQvnnzZprnguPky5dP+fLlU506dRxdCgAAAAAAAAAAAGBTGWoL6LCwMPXt21fVq1fXnj17bDJn/BXFYWFhaZ4rfuDr5eWV5rkAAAAAAAAAAAAAwNYyVAD81IEDB1SnTh117949wXVlU8vX19fQXrNmTZrnWr16dbJzAwAAAAAAAAAAAIAjZcgA2GQyKTY2VgsWLFDJkiU1efJkxcTEpGmuhg0bWq7/azabNXz4cF2+fDnV84SEhGjEiBGWeUwmkxo0aJCmmgAAAAAAAAAAAADAHjJUANy7d2+ZTCZJsgSt9+7d05AhQ1SxYkX99ttvqZ6zXbt2ln+bTCbduHFDNWrU0KZNm6yeY+PGjapdu3aCLaDjzg0AAAAAAAAAAAAAjpahAuBZs2Zp3759qlmzpmWV7dMg+Pjx42rcuLHat2+vkJAQq+esUaOG2rRpI7PZLOlJCHzlyhU1bdpUr7zyisaPH68tW7bo3LlzunnzpsLCwnTu3Dlt2bJF3377rV555RW9+eabunz5smH1b4sWLVSrVi17PRQAAAAAAAAAAAAAkGpuji4gvsqVKysoKEgLFizQ559/rmvXrllWBZvNZi1fvlzr1q3TF198oU8//VTu7u4pzjl16lTt27dPly5dkvS/1cX79+/XgQMHkh0bNzh+qkCBApo+fXpa7yIAAAAAAAAAAAAA2EWGWgEc1/vvv69Tp05p0KBBcnNzM1zHNyIiQiNGjFDp0qW1Zs2aFOfy9fXV1q1b5efnZwh0n86X3NfT46QnYXCBAgW0efNm+fn52fX+AwAAAAAAAAAAAEBqZdgAWJKyZcumH374QYcOHVKjRo0SbAt97tw5vfXWW2revLlOnz6d7FxFixbV0aNH1aNHD0lKEAQn9fX0WLPZrM6dOys4OFilSpWy7x0HAAAAAAAAAAAAgDTI0AHwU6VLl9amTZsUGBiowoULJwiCN2zYoHLlyunLL79UREREkvNkz55dP//8s/766y8NHjxY/v7+Ka4A9vPz04ABA3Ts2DEtWLBAuXLleob3HAAAAAAAAAAAAACsZzI/XQr7nHj48KHGjh2r77//Xg8fPrSEwNKT1by+vr6aMGGCOnToYNV8N2/e1OHDh3Xz5k3duXNHkuTl5aU8efKoYsWK8vb2ttt9AdLq2LFjKleunKUdHByssmXLOrAix7lx40aC12nBMdflmi2fgyrKeGLCb+jyMONjdP36deXLx2MEAAAAAAAAwLnw/jkguTm6gNTKnDmzRo8erR49emjgwIFavXq1Yavmy5cvq1OnTpo1a5amT59ueJEnJm/evGrUqNGzKB0AAAAAAAAAAAAA7Oq52AI6Mf7+/lq5cqXWr1+v4sWLJ9gW+o8//lCVKlU0YMAA3b1719HlAgAAAAAAAAAAAIDdPbcB8FNNmjRRcHCwxo0bJ09PT0MQHB0drRkzZqhEiRKaO3euo0sFAAAAAAAAAAAAALt67gNgScqUKZOGDh2qv/76S++9957hmsBms1k3b95U7969VaNGDe3du9fB1QIAAAAAAAAAAACAfThFAPxUgQIFtHDhQm3btk3ly5dPsC30vn37VLt2bX3wwQe6ceOGo8sFAAAAAAAAAAAAAJtyqgD4qbp16+rgwYOaNm2acubMaQiCY2NjFRAQoJdffllTp05VbGyso8sFAAAAAAAAAAAAAJtwygBYklxcXPTRRx/p1KlT6tmzp0wmk6T/bQt99+5dDRo0SJUqVdLvv//u2GIBAAAAAAAAAAAAwAacNgB+Kk+ePPrpp5+0Z88evfLKKwm2hQ4ODlajRo3UoUMHXbp0ydHlAgAAAAAAAAAAAECaOX0A/FTVqlW1a9cuzZs3T97e3gmC4MDAQJUuXVrjxo3To0ePHF0uAAAAAAAAAAAAAKSam6MLsEZsbKx27dqlPXv26NChQwoLC9OdO3ckSTlz5lSePHlUoUIFvfLKK6pdu7bc3JK+W926dVObNm00cuRIzZgxQ9HR0ZYQ+MGDBxo2bJjmzZunKVOm6M0333xWdxEAAAAAAAAAAAAA0i1DB8B3797V5MmTNWfOHF29ejXZYxcuXChJ8vb2Vo8ePTRo0CDlzZs30WNz5MihSZMmqVevXurfv7+2bt1quUaw2WzWmTNn1LJlS7355puaPHmyihUrZts7BgAAAAAAAAAAAAB2kGG3gP71119VtmxZjR49WleuXJHZbLZ8xRf3ttDQUH377bcqU6aMAgMDkz1HmTJltGXLFi1ZskR+fn4JtoVet26dypUrp2HDhikyMtJedxUAAAAAAAAAAAAAbCJDBsABAQFq3bq1Jfh9GsrGDWfjfiV2+82bN/X/2LvTKCurM3/Y9ykmKbAAGQJoIGoMoBEEhThEQUSFBnEKOEScOmoQxYgmarq10Rj5YxsHbI2JSZzamBajAY0jKEgQBUEREVRQBhlkkAJBEIHn/ZC3TjgUKDVx6hyua61a1t7n2fu+HxZ+8efe58wzz4z/+Z//+cZ6/fr1i1mzZsUvf/nLqF27dnrPJEniyy+/jGHDhkWbNm2+MVAGAAAAAAAAyKZqFwC/9NJL8ZOf/CS2bNmSDnQj/nXKt1mzZnHUUUdFr169omfPnnHUUUdF8+bNM04Hbx0EX3HFFfH0009/Y926devGzTffHDNnzozevXuXOg38ySefxJlnnhndu3ePmTNnVumfAQAAAAAAAEB5VKsAeOPGjTFo0KB0+Bvxz+C3efPmcdttt8WCBQtiyZIlMWHChPj73/8ezz77bEyYMCEWLVoUixYtihEjRkSrVq0yguAkSWLw4MGxYcOGnephv/32i6effjr+/ve/x3e/+91SQfC4ceOiY8eOceWVV8aaNWuq7M8CAAAAAAAAoKyqVQD85JNPxpw5c9Jha8Q/r2f+8MMPY8iQIbHPPvvscG2LFi3isssui/fffz/OO++8jO8KXrBgQTzxxBNl6qVXr17x7rvvxq9//esoLCzMCII3bdoUI0aMiDZt2pTvRQEAAAAAAACqQLUKgJ966qn076lUKn74wx/GX/7ylygsLNzpPWrXrh0PPPBAdO/ePSMEHjVqVJn7qVWrVlx33XXx/vvvR//+/UudLF62bFmZ9wQAAAAAAACoKtUqAH777bczTv9ef/316augy+qXv/xlRPwrrJ0+fXq5+2rZsmX85S9/iVdeeSUOOuigjGAZAAAAAAAAoLqoVgHw0qVLM8ZdunQp916dO3f+2r3Lo2vXrvH222/HnXfeGQ0aNKjwfgAAAAAAAACVqVoFwFu2bMkYFxSUv73ynhz+JgUFBTF48OD44IMP4sILL6ySGgAAAAAAAADlUa0C4BYtWmSM33zzzXLvNXXq1Izxt771rXLvtT1NmzaN+++/v1L3BAAAAAAAAKiIahUAH3zwwZEkSfr07i233FLuvYYNG5b+PZVKxfe///0K9wcAAAAAAABQnVWrAPjUU09N/54kSYwdOzb+/d//Pb766qud3mPz5s3x05/+NF588cVIpVKRJElERJxyyimV3i8AAAAAAABAdVKtAuDTTz89WrduHRGRDm8ffPDBaNOmTfz2t7+NTz/9dIdrly1bFr/73e+ibdu2pa5m3nvvvaNfv35V2jsAAAAAAABAttXMdgNbq1u3bowYMSJ9WrckBJ43b15cdtllcdlll0XLli1jv/32i6KiokiSJD7//PP4+OOPY9GiRRER6RO/JWtTqVTccccdUVhYmLX3AgAAAAAAANgVqlUAHBFx0kknxd133x2XXXZZpFKp9PcBlwS7ixYtisWLF2esKfksIko9P3z48Dj99NN3ResAAAAAAAAAWVWtroAucemll8bIkSOjadOmGSd6txcI7+izRo0axcMPPxxXX331rn8BAAAAAAAAgCyolgFwxD+/D/i9996La6+9Npo1axZJkmT8RESpuSRJonHjxnH11VfHrFmz4pxzzsnyWwAAAAAAAADsOtXuCuit7bXXXnHLLbfETTfdFK+99lq88cYbMX369Fi5cmUUFxdHRETDhg2jcePG0b59++jSpUscddRRUatWrSx3DgAAAAAAALDrVesAuETNmjXjmGOOiWOOOSbbrQAAAAAAAABUW9X2CmgAAAAAAAAAykYADAAAAAAAAJAnBMAAAAAAAAAAeUIADAAAAAAAAJAnBMAAAAAAAAAAeUIADAAAAAAAAJAnqk0AvHbt2my3UKny7X0AAAAAAACA6q9mthsosf/++8cNN9wQl1xySdSsWW3aKrOZM2fGtddeG126dInrr78+2+0AVJotW7bEypUrs91GzmncuHEUFFSb/98KAAAAAIA8V22S1uXLl8fgwYPjzjvvjBtvvDHOPPPMnPoP5nPnzo1f//rX8cgjj8SWLVuic+fO2W4JoFKtXLkymjVrlu02cs6yZcuiadOm2W4DAAAAAIDdRLVLWOfOnRsDBgyIAw44IO6777748ssvs93S13rrrbfijDPOiLZt28ZDDz0UmzdvznZLAAAAAAAAwG6q2gXAqVQqkiSJjz/+OAYNGhStWrWKa6+9NubMmZPt1tI2btwYjz76aHTt2jUOO+yweOKJJ2Lz5s2RJEmkUqlstwcAAAAAAADspqpNAHzBBRekf0+lUukgePny5fHf//3f0aZNm+jevXv84Q9/iBUrVuzy/pIkiXHjxsXll18eLVu2jHPPPTf+8Y9/RJIk6eC3pOfWrVvH8ccfv8t7BAAAAAAAAHZv1eY7gP/4xz/GJZdcEoMGDYqpU6emA9WIf4avERHjx4+P8ePHx8CBA+Poo4+Of/u3f4tu3brFoYceWiUnb5csWRLjxo2LsWPHxtNPP50Onkv6iYiMHvfYY4/4xS9+Eddee23sscceld4PQHXT4tr3oqB+k2y3UW1sWbsilvy/A7PdBgAAAAAAu7FqEwBHRHTp0iUmT54cDzzwQNx0002xYMGC7QbBmzdvTofBERF77rln/OAHP4gOHTrEwQcfHAcddFC0bt06GjduvFN1N2zYEJ988kl88MEHMWPGjJgxY0ZMmTIl49rp7YW+JfMFBQVx9tlnx8033xytW7eu8J8DQK4oqN8katRvmu02AAAAAACA/1+1CoAj/hmuXnjhhXHOOefEfffdF8OGDYtPP/00IwiOyAxk16xZE2PGjIkxY8Zk7FW7du1o2bJlNGzYMOrWrRt169aNGjVqxIYNG2L9+vWxbt26WLp0aaxatapUH1vvX9LX9j4/7bTT4qabbooDD3TiCwAAAAAAAMiuahcAl6hdu3YMHjw4LrroonjwwQdjxIgR8f7770dElAqDI0oHthERX375ZXz88cfpNd/0/NZ2dKV0kiRRp06dOPvss2PIkCFx0EEH7fQ7AQAAAAAAAFSlgmw38E3q1q0bAwcOjFmzZsXo0aOjd+/eUaNGjUiSpNS1zDv6iYj08zu7bmsla/bdd9+48cYbY8GCBfHHP/5R+AsAAAAAAABUK9X2BPD29OnTJ/r06RMrVqyIP//5z/H444/HG2+8EZs3b46IHZ/a3dH8jmwdELdo0SL69u0bAwYMiCOPPLL8zQMAAAAAAABUsZwKgEs0adIkBg8eHIMHD45Vq1bFiy++GC+99FK8/vrrMXv27NiyZUuF9j700EOja9eu0atXr+jQoUMldg4AAAAAAABQdXIyAN5ao0aN4owzzogzzjgjIiK++OKLePvtt+ODDz6I+fPnx/z582P58uWxbt26+OKLL2LTpk1RWFgYhYWFUVRUFN/+9rejdevWse+++0b79u2jdevWWX4jAAAAAABqaTKDAAAgAElEQVQAgPLJ+QB4W4WFhXHkkUe6rhkAAAAAAADY7RRkuwEAAAAAAAAAKocAGAAAAAAAACBPCIABAAAAAAAA8oQAGAAAAAAAACBPCIABAAAAAAAA8oQAGAAAAAAAACBPCIABAAAAAAAA8oQAGAAAAAAAACBPCIABAAAAAAAA8oQAGAAAAAAAACBPCIABAAAAAAAA8oQAGAAAAAAAACBPCIABAAAAAAAA8oQAGAAAAAAAACBPCIABAAAAAAAA8oQAGAAAAAAAACBPCIABAAAAAAAA8oQAGAAAAAAAACBPCIABAAAAAAAA8oQAGAAAAAAAACBPCIABAAAAAAAA8oQAGAAAAAAAACBPCIABAAAAAAAA8oQAGAAAAAAAACBPCIABAAAAAAAA8oQAGAAAAAAAACBPCIABAAAAAAAA8kTNbDdQYsSIEdluocwGDx6c7RaoBEuWLIk33ngjPv7441i3bl3UrVs3WrVqFYcddljsu+++2W4PAAAAAAAAdlq1CYB/9rOfRSqVynYbZVJdA+CVK1fGm2++GVOmTInJkyfHlClTYunSpRnP/Nd//VcMHTq0QnW+853vxPz58yu0R+PGjWPFihUV2qO8nn766Rg+fHi89tprkSTJdp/p0KFDDBkyJAYMGJBzfz8BAAAAAADY/VSbALjEjoK46qa6hYG33357TJkyJaZMmRJz587NdjvV2tq1a+P888+Pv/71r9/47PTp0+O8886LP/3pT/F///d/8a1vfWsXdAgAAAAAAADlU+0C4OoWrG5PdQypr7rqqmy3kBPWrl0bPXr0iDfeeKNM68aPHx8//OEPY8KECdG8efMq6g4AAAAAAAAqptoFwJUdrpY3UN5RH7kQUGfLWWedFa1atSrTmnr16lVRN9t34YUXlgp/W7VqFUOGDInjjjsu9tlnn1iyZElMnDgxbr/99pg1a1b6uTlz5sTpp58e48ePj5o1q92/OgAAAAAAAFB9AuD27dtXOFxdsWJFLFq0qNQ+W4e59erVi6KiomjQoEEkSRJr1qyJNWvWxLp169LPpFKp9B5JkkQqlYq99947GjduXKH+dqX69etHp06donPnztG5c+c488wzq7zmxRdfHN26davyOuX19NNPx8iRIzPmunbtGqNGjYoGDRqk5xo2bBjt2rWLAQMGxIABAzLWvPbaa3HffffFZZddtsv6BgAAAAAAgJ1VbQLgt99+u0LrH3zwwbjyyiszgtuIiP333z/OOuusOOqoo6JDhw47vL536dKlMX369Jg4cWI89thjMXfu3HQQnCRJrF27Nn71q1/FeeedV6E+q0pJ0Fvy065duygoKEh/visC4OosSZK4/vrrM+ZatWoVzzzzTNSvX3+7a+rUqROPPfZYfPTRRzF16tT0/K9//eu48MILo7CwsEp7BgAAAAAAgLKqNgFwRVxxxRXxP//zP+lxkiTRoUOH+M1vfhPdu3ffqT2aN28ezZs3jxNPPDFuuummePnll+Oqq66K6dOnRyqViuLi4rjwwgvjrbfeijvvvLOqXqXcJk+enO0WqrVRo0bF9OnTM+buuuuuHYa/JWrUqBH3339/dOrUKT23dOnSuP/+++OKK66okl4BAAAAAACgvAq++ZHq7dprr4277747kiRJ/9xwww0xbdq0nQ5/t6d79+4xbdq0uP7669PXQCdJEnfffXdcd911lfgG7AqPP/54xvg73/lO9O3bd6fWduzYMX74wx9mzG17lTQAAAAAAABUBzkdAL/00ktx6623pq99TqVScccdd8TQoUMr/H3CJfvdeOONceedd2aEwLfeemuMHTu2wvuza3z11Vfx3HPPZcwNGDAg44rsb3L++ednjCdNmhTLli2rjPYAAAAAAACg0uR0AHzVVVelf0+lUtG3b98quZZ38ODB0bdv34wQeMiQIZVeh6oxadKkKC4uzpjr1q1bmfbY9vktW7bE888/X8HOAAAAAAAAoHLlbAA8bty4ePfdd9OBbETE8OHDq6ze//t//y9j/O6778Yrr7xSZfWoPO+8807GuEaNGvGDH/ygTHvsv//+8a1vfStjbsaMGRXuDQAAAAAAACpTzWw3UF6jRo3KGHfs2DG+973vVVm9tm3bRqdOnWLatGnpudGjR8exxx5bZTVzzebNm2Pq1Knx1ltvxfLly2PDhg2x1157RePGjaNDhw7x/e9/v1Ku5i6r2bNnZ4xbtWoV9erVK/M+7dq1i08//XSH+wIAAAAAAEC25WwAPGXKlIiI9LXMHTp0qPKahxxySEybNi0dYpb0wD/17NkzNm3atMPP99prrzj55JPj5z//ebRr126X9TVr1qyMcatWrcq1z7e//e2MsQAYAAAAAACA6iZnr4CeM2dOxmnSli1bVnnNFi1apH9PkiTmzJlT5TVzydeFvxERn332WTzwwANx0EEHxYUXXhhffPHFLulr8eLFGeNtg9ydtW1wvGjRonL3BAAAAAAAAFUhZwPg1atXZ4zXrVtX5TW3rbFtD+ycJEnigQceiC5dusSCBQuqvN7atWszxkVFReXaZ9t169evjy1btpS7LwAAAAAAAKhsOXsFdJIkGeOFCxdWec1ta2zbw+6qffv2cdJJJ0WXLl3ioIMOiqZNm0ZhYWEUFxfHggULYsKECfHwww9nfH9yRMTMmTOjT58+MXHixNhzzz2rrL9tA+C6deuWa5/trVu7dm25A+USy5Yti+XLl5dpjdPnAAAAAAAAbE/OBsB77bVXfPrpp5FKpSJJkhg7dmx89dVXUatWrSqpt3Hjxhg7dmzGtdN77bVXldTKFRdeeGGcdtpp8f3vf3+7nzdp0iSaNGkSnTp1iiuuuCJGjhwZF110UcbJ6RkzZsSgQYPi4YcfrrI+tz25vccee5Rrn6oKgO+999648cYbK7QHAAAAAAAAROTwFdBt27bNOIG7evXqePzxx6us3uOPPx7FxcUR8c+Tv6lUKtq0aVNl9XLBDTfcsMPwd3v69esXEydOjAYNGmTMP/roozFjxozKbm+Htg7xy2J7J77LuxcAAAAAAABUhZwNgLt165b+veQU8JAhQ2LFihWVXmv58uUxZMiQUmFf165dK71WvjvooIPiD3/4Q8bcli1b4o477qiymvXq1csYr1+/vlz7bNiw4Rv3BgAAAAAAgGzK2SugzzjjjFLX5i5fvjx69uwZzz77bDRr1qxS6ixbtix69eoVK1asKBUAn3XWWZVSY3fzox/9KA499NCYOnVqeu6FF16osnr169dPn96OKH8AvL119evXL3dfJS699NLo169fmdbMmTMnTjnllArXBgAAAAAAIL/kbADcpk2b6Nu3b4waNSpSqVQ6nJ02bVocfvjhce+990bPnj0rVOO5556LQYMGxbx589L7l1z/3Ldv393+CuiKOP300zMC4MWLF8eHH34YBxxwQKXX2jakXbNmTbn22XZd3bp1o6Cg4ofomzVrVmn/wwIAAAAAAAC7t5y9Ajoi4vbbb9/uCcx58+ZF79694/jjj4+RI0eW6cTn+vXr4/HHH4/jjz8++vTpE/PmzSv1TL169eI3v/lNRVrf7R1xxBGl5hYtWlQltVq2bJkxXrhwYbn22XbdtvsCAAAAAABAtuXsCeCIiH333Tf++Mc/xllnnRVJkkREZJzUffnll+Pll1+OunXrxiGHHBIdOnSIAw44IIqKiqKoqCgi/nmqc82aNfHhhx/GO++8E2+99VY6MC457VsiSZIoKCiIP/zhD7Hffvvt4rfNL9s78bp8+fIqqdW2bdt4+eWX0+MFCxaUa59t17Vr165CfQEAAAAAAEBly+kAOCKiX79+sXr16hg4cGBs2bIlPZ9KpdKh8BdffBGTJk2KSZMmfeN+JWtK9th6vqCgIO69997o379/Jb7B7mnrP+cS237HcmXZNqhdsGBBrFu3LurVq1emfWbPnp0xbtu2bYV7AwAAAAAAgMqU01dAl/jJT34SzzzzTLRs2bJUgFvykyTJTv1svaZEkiTRokWLePrpp+Piiy/OxivmnU8//bTUXNOmTaukVvv27TPGmzdvjsmTJ5dpj7lz58bSpUsz5g4++OAK9wYAAAAAAACVKS8C4IiIE088MWbNmhWDBg3KOP1bYutg9+t+tlYSCA8cODBmzZoVvXr12pWvlNe2dxq7RYsWVVLriCOOiIYNG2bMjRs3rkx7jB8/PmNcUFAQPXv2rGhrAAAAAAAAUKnyJgCOiKhfv37cfffd8cEHH8Q111wTzZs3zzjd+022frZZs2bxi1/8ImbPnh333HNP+juDqbgkSeKJJ57ImGvRokV873vfq5J6tWrVKhXeP/LIIzv1d6LEQw89lDE+4ogjtvs9xgAAAAAAAJBNOf8dwNuz3377xbBhw+Lmm2+O119/PSZPnhxTpkyJd955J1auXBnFxcXx5ZdfRkREnTp1okGDBtG4ceM4+OCDo0uXLtGlS5c44ogjokaNGll+k/z0l7/8Jd5+++2MuRNPPLFKa/bv3z8ee+yx9Pjjjz+Op59+Ovr27fuNa6dPnx6vvvpqxly/fv0qvUcAAAAAAACoqLwMgEvUqFEjjjrqqDjqqKNKfbZx48ZIkiTq1KmThc5y24oVK2LPPfcs15/d9OnT45JLLsmYS6VS8bOf/Wyn1nfr1q3Udcw7c5L35JNPjg4dOsT06dPTc4MHD47u3btH/fr1d7hu8+bNcdFFF2XMNW/evNQcAAAAAAAAVAd5dQV0WdSuXVv4W07/+Mc/Yv/994+77rorPvvss51akyRJPProo3H00UfH559/nvHZj3/84+jQoUNVtJqWSqXiV7/6Vcbc/Pnzo2/fvrFmzZrtrtm4cWOcc845MWXKlIz5//iP/4jCwsIq6xUAAAAAAADKK69PAO9Opk6dGiNHjtzp58eMGRMbNmzY7meNGjWKa6655mvXL1q0KH72s5/F1VdfHccdd1x07do1OnbsGPvvv380aNAgCgsLY/Xq1bFgwYKYMGFCPPzwwzFz5sxS+3To0CHuueeene67Ik466aQ4/fTT469//Wt67pVXXon27dvHVVddFccdd1y0bNkyPv3005g4cWL85je/iffeey9jjyOOOCJ++tOf7pJ+AQAAAAAAoKwEwHlixowZMXz48J1+fuLEiTFx4sTtfta6detvDIBLbNq0KV544YV44YUXdrp2iQ4dOsSzzz4bRUVFZV5bXg888EAsXLgwJk+enJ6bP39+DB48+BvX7r///vHXv/41atb0rw0AAAAAAADV0257BTTZU6tWrbjqqqti8uTJ0bJly11ae88994wxY8bEqaeeWqZ1Rx99dPzjH/+IFi1aVFFnAAAAAAAAUHECYMqsa9eu8ac//SkGDBgQBxxwQBQU7NxfozZt2sR//ud/xrx58+K2226L2rVrV3Gn27fnnnvGk08+GaNGjYojjzzya59t3759PPjggzF+/Pho3rz5LuoQAAAAAAAAyme3uMt2wYIFMW3atFi+fHmsWrUqVq1aFV9++WVERPTu3TuOO+64LHdYceeff36cf/75u6RWo0aN4oILLogLLrggIiLWrl0b77//fixYsCAWL14c69ati40bN0b9+vWjUaNG0bx58+jcuXPstddeFa49bty4Cu9Rom/fvtG3b99YvHhxvP766zFv3rxYt25d1K1bN7797W9H586dY7/99qu0egAAAAAAAFDV8jYAfumll+JPf/pTTJgwIZYsWbLD55o0afK1AfCCBQvi9ddfz5j77ne/G506daq0XnNd/fr149BDD41DDz00262US8uWLeO0007LdhsAAAAAAABQYXkXAD/66KNxyy23xOzZsyMiIkmSHT6bSqW+cb/CwsK44IILYsOGDem5tm3bxsyZMyveLAAAAAAAAEAlypvvAF61alX86Ec/inPPPTdmzZoVSZJEkiSRSqW2+7OzmjRpEuedd156vyRJYvbs2TF58uQqfBsAAAAAAACAssuLAPiDDz6IQw45JJ566qn0id9tg96tA9yyuvLKK9N7lvjf//3fCnYNAAAAAAAAULlyPgBesmRJdO/ePRYuXJhx4jfiX6FvgwYN4tBDD40TTjihXDUOOOCA6NixY3r/JEni+eefr8zXAAAAAAAAAKiwnA+AzzzzzFi8eHGp4Ld+/foxZMiQmD59eqxcuTKmTJlSodC2X79+GeO5c+fGggULKtQ7AAAAAAAAQGXK6QD4wQcfjAkTJpQ68dujR4/48MMP47bbbouDDz64TN/5uyO9e/cuNTdu3LgK7wsAAAAAAABQWXI2AE6SJG6++eaM8DeVSkX//v3jhRdeiGbNmlVqvQMPPDD22GOPjLmZM2dWag0AAAAAAACAisjZAPjVV1+Njz76KCL+Gf5GRBx00EHxyCOPVMqJ323VqFEjDj744HStiIjZs2dXeh0AAAAAAACA8srZAPi5557LGKdSqRg+fHjUqlWrymoecMAB6VpJksTcuXOrrBYAAAAAAABAWeVsADx58uSMcdOmTaNXr15VWrNhw4YZ4+Li4iqtBwAAAAAAAFAWORsAz5kzJ30SN5VKxbHHHlvlNbcNgNesWVPlNQEAAAAAAAB2Vs4GwJ999lnGeO+9967ymjVq1MgYr1+/vsprAgAAAAAAAOysnA2Av/zyy4xx/fr1q7zmtqFznTp1qrwmAAAAAAAAwM7K2QC4bt26GeNVq1ZVec1ly5ZljBs0aFDlNQEAAAAAAAB2Vs4GwE2aNMkYf/LJJ1Ve84033ohUKpUef/vb367ymgAAAAAAAAA7K2cD4P322y+SJIlUKhVJksQ//vGPKq03f/78WLBgQUREuu6BBx5YpTUBAAAAAAAAyiJnA+DDDjssY7xixYp47bXXqqze73//+1JzP/jBD6qsHgAAAAAAAEBZ5WwAfNxxx5WaGzZsWJXUKi4ujt/+9rcZ1z9HRPTq1atK6gEAAAAAAACUR84GwN27d09/D3DJNdDPPvtsPPbYY5Ve69xzz43i4uKI+Nf1z4cffni0atWq0msBAAAAAAAAlFfOBsA1atSISy+9NJIkiYh/hcAXX3xxjBs3rlJqJEkSgwcPjmeeeabU6d/BgwdXSg0AAAAAAACAypKzAXBExJVXXhnNmjVLj1OpVKxbty569uwZI0aMiC1btpR77/nz50evXr3innvuSYe/Jad/DznkkDjjjDMq3D8AAAAAAABAZcrpALhBgwYxYsSI9CngkoB248aNceWVV8aBBx4Y99xzTyxZsmSn9kuSJCZOnBgDBw6MNm3axEsvvZTeu0TNmjXjd7/7XaW/CwAAAAAAAEBF1cx2AxXVv3//eOONN+KOO+5In9QtuQ76gw8+iMGDB8cVV1wRrVq1irZt25Za//LLL8fChQvjo48+imnTpsVnn30WEZFxtXTJOJVKxW233RaHHXbYLno7AAAAAAAAgJ2X8wFwRMRvfvObWLduXfz+97/PCIEj/hncJkkS8+bNi/nz56fnSv75yiuvxCuvvJIxv/X6rf3yl7+Myy+/vErfBQAAAAAAAKC8cvoK6K3dd999ceutt0aNGjUy5lOpVPpn2+ucI/4VEJec8C352frzmjVrxogRI+JXv/pVlb8HAAAAAAAAQHnlTQAcEXH11VfHlClT4vDDD0+HulvbOuDd3s/WStYfdthh8frrr8dll122K18FAAAAAAAAoMzyKgCOiOjQoUNMnDgxnn/++ejbt2/UqFEj45Tvjmz7zDHHHBNPPPFETJ48OTp27Lir2gcAAAAAAAAot7z4DuDtOeGEE+KEE06INWvWxNixY2PSpEnxzjvvxPz582Pp0qXxxRdfxObNm2OPPfaIRo0aRatWreLAAw+MH/zgB/Fv//Zv0bJly2y/AgAAAAAAAECZ5G0AXKKoqChOPfXUOPXUU7PdCgAAAAAAAECVyrsroAEAAAAAAAB2VwJgAAAAAAAAgDwhAAYAAAAAAADIEwJgAAAAAAAAgDwhAAYAAAAAAADIEwJgAAAAAAAAgDwhAAYAAAAAAADIEzWz3UBVWLt2bUyZMiXeeuutmDlzZnz22WexevXq+Pzzz2Pz5s2VUiOVSsXUqVMrZS8AAAAAAACAypBXAfD48ePj97//ffztb3+LDRs2lPo8SZIK10ilUpEkSaRSqQrvBQAAAAAAAFCZ8iIAXr58eVx++eUxcuTIiKicoBcAAAAAAAAg1+R8ADxv3rw45phjYtGiReng1+lcAAAAAAAAYHeU0wHwqlWrokePHvHJJ59ExPaDX6eBAQAAAAAAgN1FTgfAQ4cOjY8++igd/JZ8N29J6Nu4cePo1KlTtGvXLho1ahRFRUVRUFCQzZYBAAAAAAAAqkzOBsALFy6M3/72txmnfkvC3yOPPDKuv/766NGjR9SoUSOLXQIAAAAAAADsOjkbAI8ePTo2bdpU6vTv0KFD44YbbshydwAAAAAAAAC7Xs7eh/z888+nfy8Jf//93/9d+AsAAAAAAADstnI2AJ4zZ07G9c+1atWKYcOGZbEjAAAAAAAAgOzK2QB4+fLlEfGv079HH310NG7cOMtdAQAAAAAAAGRPzgbAa9asyRgfeOCBWeoEAAAAAAAAoHrI2QC4fv36GeO99torS50AAAAAAAAAVA85GwC3bt06Y7xq1aosdQIAAAAAAABQPeRsANyxY8f09/9GRCxatCjLHQEAAAAAAABkV84GwL17907/niRJjB8/PovdAAAAAAAAAGRfzgbAJ510UrRo0SI9XrlyZYwZMyaLHQEAAAAAAABkV84GwLVr147//M//TF8DnSRJXHfdddluCwAAAAAAACBrcjYAjogYOHBgHHvssekQeNq0afGzn/0s220BAAAAAAAAZEVOB8AREU888US0bds2kiSJJEni7rvvjsGDB8fGjRuz3RoAAAAAAADALpXzAXCjRo1iwoQJ0blz54iISJIk7rnnnujUqVP83//9X2zevDnLHQIAAAAAAADsGjWz3UBFjB49Ov371VdfHTfeeGO89957kSRJvPfee3H22WfH5ZdfHkcffXR07tw5mjVrFg0bNoyaNSvntfv27Vsp+wAAAAAAAABUhpwOgE855ZRIpVKl5lOpVPpK6BUrVsTf/va3+Nvf/laptVOpVGzatKlS9wQAAAAAAACoiJwOgEskSVJqvHUwvO3nAAAAAAAAAPkoLwLg7Z0CLsvnZSVQBgAAAAAAAKqjgmw3AAAAAAAAAEDlyOkTwEVFRZV+uhcAAAAAAAAgV+V0AFxcXJztFgAAAAAAAACqDVdAAwAAAAAAAOQJATAAAAAAAABAnhAAAwAAAAAAAOQJATAAAAAAAABAnhAAAwAAAAAAAOQJATAAAAAAAABAnhAAAwAAAAAAAOQJATAAAAAAAABAnhAAAwAAAAAAAOSJmtluoMTo0aN3+Fnfvn3LvGZX2FFfAAAAAAAAANlQbQLgU045JVKpVKn5VCoVmzZtKtOaXeHr+gIAAAAAAADIhmoTAJdIkmSXrAEAAAAAAADIN9UuAN76RO/OBru7+hSwwBkAAAAAAACojgqy3QAAAAAAAAAAlaPanAAuKioq80ne8qwBAAAAAAAAyFfVJgAuLi7eJWsAAAAAAAAA8pUroAEAAAAAAADyhAAYAAAAAAAAIE8IgAEAAAAAAADyhAAYAAAAAAAAIE8IgAEAAAAAAADyhAAYAAAAAAAAIE8IgAEAAAAAAADyhAAYAAAAAAAAIE/UzHYD5I9ly5bFjBkz4uOPP45Vq1bFpk2bolGjRtG0adPo1KlT7LvvvtluEQAAAAAAAPLabhEAL168OFatWhXFxcWxevXq2LRpU6Xs26NHjygsLKyUvSrTypUr480334wpU6bE5MmTY8qUKbF06dKMZ/7rv/4rhg4dWqE6xcXF8fe//z3GjBkTY8eOjYULF37t8y1atIgBAwbEoEGDolWrVhWqXR6pVKrCexx66KHx5ptvVkI3AAAAAAAAUPnyMgD+8MMP49FHH43XX389Jk+eHKtXr66SOrNmzYrvfe97VbJ3Wd1+++0xZcqUmDJlSsydO7dKa3300UdxxRVXxIsvvhgbN27c6XVLliyJW2+9Ne6444646aab4he/+EUUFLiFHAAAAAAAACpLXgXAEydOjF/96lcxZsyYSJIkIiL9z8pWGadJK9NVV121y2p98MEH8cwzz5R7/VdffRXXXXddTJw4MZ588smoVatWJXYHAAAAAAAAu6+8CICTJImhQ4fGLbfcElu2bMkIfasiqK2qUDmX7b333nHCCSdEt27dokOHDtGsWbOoV69erFixIt5444146KGH4oUXXshY88wzz8RPfvKTeOihh7LS809/+tNo0KBBmdbss88+VdQNAAAAAAAAVFxeBMBnnnlmPPHEE+lgdkeh77bB7deFw9sLeavbqd+vU79+/ejUqVN07tw5OnfuHGeeeWal10ilUnHSSSfFxRdfHD179owaNWqUeqaoqCj222+/OOuss2LUqFExYMCA+Pzzz9OfP/zww3H22WfHiSeeWOn9fZNrrrkmvvOd7+zyugAAAAAAAFBVcj4AvvHGG2PkyJERkRnQbh3g1qpVK2rUqBEbNmyIVCoVSZJEKpWKoqKi2LhxY6xfvz5jz1Qqld6r5NnCwsKoWTPzj2t7gWe2lAS9JT/t2rXL+H7dygyAU6lU9OzZM2655Zbo2LHjTq87+eST48knn4wTTzwxtmzZkp6/4YYbshIAAwAAAAAAQL7J6QB45syZcdNNN5U6mZskSfTo0SMuuuiiOPbYY6NJkybx61//Oq6//vqM51atWhURERs3bozly5fHtGnT4rXXXovHHnssFixYkA6CkySJVq1axSOPPBKdOnXaZe9XFpMnT95ltXr06FHuwLZHjx4xYMCAjGufp0yZEosXL46WLVtWVosAAAAAAACwWyr45keqr1tuuSXjpG+SJHqOkfIAACAASURBVFGzZs14+OGH48UXX4x+/fpFkyZNvnGf2rVrx9577x0nnXRSDBs2LD766KN4/PHHY5999kmfAJ49e3YcffTR8dRTT1XlK+WEip58Pu+88zLGSZLEa6+9VqE9AQAAAAAAgBwOgJcsWRKPP/54qauaH3jggTjnnHMqtHdBQUH86Ec/ihkzZsSpp56aDpnXr18fZ5xxRrz44osV7n931qFDh1JzS5YsyUInAAAAAAAAkF9yNgAeN25cbN68OSL+Ff726dMnzj777EqrUVRUFI8//nicfPLJ6RqbNm2Ks846K5YtW1ZpdXY3devWLTW3bt26LHQCAAAAAAAA+SVnA+BXX3211NxVV11V6XVq1KgRjz76aOyzzz7pueLi4rjuuusqvdbuYsGCBaXmmjVrloVOAAAAAAAAIL/UzHYD5fX2229njJs1axbHHHNMldQqLCyMW265Jc4999xIpVKRJEk8+uijMWzYMMFlOYwbN67U3P7777/L+/jyyy9j4sSJ8e6778by5ctj8+bN0bhx42jSpEkceuihccABB+zyngAAAAAAAKAicjYAXrFiRTqMTaVScdhhh5V5j/Xr12/3OuLt6devXwwcODC++OKLiIj46quvYtSoUXHRRReVue7u7qGHHsoYFxUVxZFHHrnL+/j+978fmzZt2uHnLVq0iH79+sVVV10VrVq12oWdAQAAAAAAQPnk7BXQn332WcZ43333/drna9YsnXWvX79+p+vVqVMnunXrFkmSpOdeeeWVnV7PP73wwgsxadKkjLnTTjstatWqtct7+brwNyJiyZIlMWLEiPjud78b11577Tc+DwAAAAAAANmWswHw559/njFu2LDh1z6/5557lppbvXp1mWq2bt06IiJ98vi9994r0/rd3eeffx4//elPM+Zq1apV7b9P+auvvorhw4dH9+7dy/x3BgAAAAAAAHalnL0CurCwMCMETqVSX/v89gLghQsXfuPJ4a01adIkY/zJJ5/s9FoiLrnkkpg3b17G3JAhQ+J73/veLuuhoKAgunTpEr17947DDjss2rVrF40bN446derEqlWr4qOPPorx48fHH//4x/jwww8z1k6YMCF+9KMfxXPPPbfdE+XltWzZsli+fHmZ1syZM6fS6gMAAAAAAJA/cjYALioqygiAi4uLv/b57Z0Qnj9/fplqlnz/b4m1a9eWaf3u7LbbbovHHnssY659+/Zx44037rIefv7zn8fAgQN3GPo3a9YsmjVrFocffnj8/Oc/j/vuuy+GDBkSX375ZfqZMWPGxE033RQ33XRTpfV177337tI/BwAAAAAAAPJXzl4B3bx584zv4121atXXPr+9U6avv/56mWrOnTs3Y1yZp0Dz2d/+9re45pprMuaKiopi5MiRUadOnV3Wx6233rrTJ74LCgri0ksvjeeee67U9xPffvvtsWzZsqpoEQAAAAAAACokZwPgdu3aRcS/vo939uzZX/v8AQccEHXr1s1Y88orr+x0va+++ipeffXVjKumGzduXI7Ody+vvvpqnHXWWbFly5b0XO3ateOJJ57YpVc/l9exxx4bt9xyS8bcunXr4r777stSRwAAAAAAALBjOXuEtSQALjFz5sxIkmSH3wVcUFAQHTp0iNdffz39zPvvvx/jx4+Prl27fmO9Rx55JFauXJkOj1OpVLRu3briL5LH3nrrrejbt29s2LAhPVdQUBAPP/xwHH/88VnsrGwuv/zyuOOOO2Lx4sXpueeffz5uuOGGStn/0ksvjX79+pVpzZw5c+KUU06plPoAAAAAAADkj5wNgI888siM8YYNG+Kdd96JDh067HDN6aefnnHtc5IkMXjw4Jg0aVIUFhbucN2HH34YV199dalw+Zhjjiln9/nv/fffjxNPPDFWr16dMX/vvffGGWeckaWuyqdOnTrRp0+f+P3vf5+emzx5cqxfvz59qrwiSr57GAAAAAAAACoqZ6+APvzww0uFb0899dTXrjnrrLOioOCfr1wS5r777rvRrVu3mDFjxnbXPPnkk3H00UdHcXFxqc/69OlTntbz3rx586JHjx6xfPnyjPnhw4fHJZdckqWuKuaII47IGG/evDk+/fTTLHUDAAAAAAAA25ezAXDt2rXj6KOPTl/HnCRJPPnkk1+7pmXLlnHOOedEkiTpuSRJ4s0334xDDjkkDjnkkDjnnHPi8ssvj7PPPjtat24d/fr1i2XLlqUD45J6P/zhD+Pwww+v0nfMRYsXL44ePXrEJ598kjH/H//xH/GLX/wiS11V3PZO6G4bcAMAAAAAAEC25ewV0BER/fv3jxdffDE9njlzZkyaNKnUac2t/fd//3eMHj06Vq9enRHqJkkS77zzTsZJ4JKgeNurn2vVqhW33XZbZb5KXlixYkUcf/zxMXfu3Iz5yy+/PG6++eYsdVU5tv6fBkrs6PumAQAAAAAAIFty9gRwRMRpp50WtWvXTodzSZLEsGHDvnZN06ZN489//nPUrl07PZdKpUqFwSUnfbcO+Urq3HXXXdG5c+fKfp2ctnr16jjhhBPivffey5i/4IIL4q677spSV5Vne9c9N23aNAudAAAAAAAAwI7l9Anghg0bxujRo2PVqlXpuZLv+P06PXv2jCeffDJ+/OMfR3FxcTrk/boTnUmSRJ06deJ3v/tdnHvuuRVvPo+sW7cuevXqFW+99VbGfP/+/eP+++/Pi5OykyZNyhjXqFFju9dCAwAAAAAAQDbldAAcEXHCCSeUa12vXr3iww8/jF/+8pfx6KOPxhdffLHDZwsKCqJ///4xdOjQaNOmTXlbzUtffvllnHzyyaUC0j59+sT//u//Ro0aNbLUWeVZv359PPPMMxlznTt3jrp162apIwAAAAAAANi+nA+AK6Jx48bxu9/9Lu66664YO3ZsvPnmm/Hpp5/GypUro169etG0adPo1KlT9OjRIxo3bpztdqudTZs2Rf/+/WPs2LEZ8927d4+RI0dGrVq1stRZ5brzzjtj6dKlGXM9e/bMUjcAAAAAAACwY7t1AFxijz32iN69e0fv3r2z3UrO2LJlS5x33nkxevTojPkjjzwyRo8eHXvssUeV1O3WrVuMHz8+Y67ku5l3ZOnSpdG0adNynUZ+8cUX44YbbsiYKywsjEsuuaTMewEAAAAAAEBV++YvzIXtGDRoUPz5z3/OmOvUqVM8++yzUa9evSx1tX1/+ctf4sADD4wHHngg1q5du1NrNm3aFHfccUf06dMnNm3alPHZkCFDonnz5lXRKgAAAAAAAFSIE8B5YurUqTFy5Midfn7MmDGxYcOG7X7WqFGjuOaaa3a49v7774/77ruv1HynTp1i2LBhO93D1vbbb7+4+OKLy7V2Z3zwwQdx4YUXxqBBg6Jnz55x1FFHRceOHaN169bRoEGD2GOPPWLVqlXx0Ucfxbhx4+KBBx6I+fPnl9rnhBNOKHUiGAAAAAAAAKoLAXCemDFjRgwfPnynn584cWJMnDhxu5+1bt36awPgRYsWbXf+D3/4w07X31bXrl2rNAAusX79+njqqafiqaeeKvPa7t27xxNPPJE3320MAAAAAABA/nEFNHyDwsLCGD58eLz00kux5557ZrsdAAAAAAAA2CEngMl7Z5xxRtSpUydeeeWVmDx58navdt5WQUFBHHzwwfHjH/84fvKTn0SjRo12QacAAAAAAABQMQLgPHH++efH+eefv0tqDR06NIYOHbpLam1r3LhxZV7TokWLGDhwYAwcODAiIoqLi2P27NmxcOHCWLp0aaxbty42bdoURUVF0ahRo9hnn33isMMOc9oXAAAAAACAnCMAZrfTsGHDOPzww+Pwww/PdisAAAAAAABQqapNADx69Ohst1Bmffv2zXYLAAAAAAAAAGnVJgA+5ZRTIpVKZbuNnZZKpWLTpk3ZbgMAAAAAAAAgrdoEwCWSJMl2CwAAAAAAAAA5qdoFwLlwClhIDQAAAAAAAFRHBdluAAAAAAAAAIDKUW1OABcVFeXE6V8AAAAAAACA6qraBMDFxcXZbgEAAAAAAAAgp7kCGgAAAAAAACBPCIABAAAAAAAA8oQAGAAAAAAAACBPCIABAAAAAAAA8oQAGAAAAAAAACBPCIABAAAAAAAA8oQAGAAAAAAAACBP1Mx2A7vKpk2bYunSpbFq1aooLi6OtWvXRv369aNhw4bRsGHDaN68edSqVSvbbQIAAAAAAACUW94GwF999VWMGjUqxo8fH1OmTInp06fHxo0bd/h8rVq1on379tGlS5fo2rVrnHzyyVG7du1d2DEAAAAAAABAxeRdADxv3ry4++6745FHHomVK1dGRESSJN+4buPGjfHmm2/G1KlT47e//W00+v/Yu/Moq6ozb8DvLSgmGcOggoLacUQtggoRlKgxajSoUTDEqEEytbSmPyNGjQloTNQYgybdarTbqLEhitMCEQc0ikFBEMFCgcigkYAyFLOCUMX5/nBVhVtVYA331uWWz7NWLdi7zt7vexz++q29T4cOcdFFF8Wll14aBxxwQLbbBgAAAAAAAKi3RvMN4LKysrj55pujZ8+ecfvtt8fq1asjSZKK8DeVSn3mT0RUrFmzZk38/ve/j8MPPzxuuummKCsry+XrAQAAAAAAAHymRhEAv/fee3H00UfHtddeG5s3b44kSaoNdz9L5TVJksSWLVvi5z//eRx11FHx7rvvZvlNAAAAAAAAAOou7wPgefPmxZe//OUoLi5OC353VH6qtyY/O9oxCC4uLo5jjz025s2b15CvBwAAAAAAAFBjef0N4JUrV8Ypp5wSK1eu3Gnw27Rp0+jfv38UFRXFkUceGV26dIm2bdvGHnvsER999FFs2LAhVq5cGcXFxfHmm2/GK6+8EqWlpWl7lf995cqVceqpp8asWbOiS5cuDfquAAAAAAAAAJ8lrwPgYcOGxfLly6sNfnv06BE//elP47zzzouOHTvWeM+SkpJ4+OGH49Zbb4333nuvyt7Lly+PYcOGxcSJEzPyDgAAAAAAAACZkrdXQE+ePDkmTZqUFtCWXwF93XXXxYIFC+KSSy6pVfgbEdGxY8cYPnx4zJ8/P0aNGlXlJHCSJPH000/H5MmTM/YuAAAAAAAAAJmQtwHw7bffnjZOkiSaN28eDz/8cIwcOTKaN29er/2bN28eo0aNiocffjiaNWtW5fe33XZbvfYHAAAAAAAAyLS8DIA3btwYkydPrjidW37y98Ybb4xzzz03o7XOPffc+PWvfx1JkkTEv04Bv/DCC7Fx48aM1gIAAAAAAACoj7wMgGfOnBmlpaURERXBbFFRUVx++eVZqXf55ZdHUVFRRa2IiNLS0pgxY0ZW6gEAAAAAAADURV4GwAsXLkwbp1Kp+O53v5u1eqlUKoYOHVplftGiRVmrCQAAAAAAAFBbeRkAr1+/vsrcySefnNWa1e2/YcOGrNYEAAAAAAAAqI28DIALCwurzO2zzz5ZrVnd/k2bNs1qTQAAAAAAAIDayMsAuEOHDlXmmjdvntWazZo1q1EfAAAAAAAAALmSlwHwwQcfXGXun//8Z1ZrLl++vMrcQQcdlNWaAAAAAAAAALWRlwFwr169okWLFmlzr7/+elZrzpw5M23cokWL6N27d1ZrAgAAAAAAANRGXgbALVu2jFNOOSWSJIlUKhUREWPGjMlqzf/7v/+r+HsqlYpTTjmlSggNAAAAAAAAkEt5GQBHRFx++eUVf0+SJCZNmhQvvPBCVmpNnjw5Jk2aFKlUKpIkqVIfAAAAAAAAYHeQtwHwV77ylRg0aFDFKeAkSWLo0KHx7rvvZrTOkiVLYtiwYRUnjVOpVAwaNCgGDBiQ0ToAAAAAAAAA9ZW3AXBExN133x2HHHJIRQi8bNmy6NevX0ybNi0j+7/66qvRv3//WL58eUR8etL4kEMOibvvvjsj+wMAAAAAAABkUl4HwB06dIjJkyfHkUceWRECr1ixIo4//vi46KKLYuHChXXad+HChXHRRRfFgAEDYsWKFZEkSSRJEkVFRTF58uRo3759ht8EAAAAAAAAoP6a5rqB+urWrVu8+uqrceWVV8Yf//jHSKVSsX379hgzZkyMGTMmevfuHWeeeWb06tUrjjjiiNhzzz2jZcuWFes3b94cK1eujOLi4pgzZ05MmDAh3njjjYiIiu/9FhQUxL//+7/HLbfcEq1atcrJewIAAAAAAAB8lrwOgL/whS+kjcu/BVz+Z0TErFmzKgLdck2aNImWLVvG5s2bo6ysLO135esq7zd27NgYO3ZsnXtNpVJRUlJS5/UAAAAAAAAAnyWvA+B169aljXcMbFOpVERExfXNOyotLY2NGzfudN/yteW2b99epVZtVd4TAAAAAAAAINPyOgCOSA9Wy4Peyqd4a6tyYFweKtdV5f0AAAAAAAAAsiHvA+AdZeqUrdO6AAAAAAAAQD7K+wDY6VoAAAAAAACAT+V1APzEE0/kugUAAAAAAACA3UZeB8BnnXVWrlsAAAAAAAAA2G0U5LoBAAAAAAAAADJDAAwAAAAAAADQSAiAAQAAAAAAABoJATAAAAAAAABAIyEABgAAAAAAAGgkBMB19Nprr+W6BQAAAAAAAIA0eR0ADx8+PD755JMGr3vLLbfEgAEDGrwuAAAAAAAAwK7kdQD8xz/+Mb785S/HO++80yD1Vq9eHaeffnpcffXVUVpa2iA1AQAAAAAAAGoqrwPgiIji4uI4+uij48EHH8xqnZdeeil69eoVzz77bFbrAAAAAAAAANRV3gfAERGbNm2KoUOHxtChQ+Pjjz/O6N5JksR1110XX/va12L58uUZ3RsAAAAAAAAgkxpFAJxKpSJJknjwwQfj6KOPjrlz52Zk3w8++CBOPPHEuOGGG6KsrCxSqVRG9gUAAAAAAADIhkYRAEf8KwResGBB9O3bN+6+++567ff0009HUVFR/O1vf4skSaqEv127dq3X/gAAAAAAAACZltcB8PDhwyNJkopxKpWKVCoVW7ZsieHDh8e3vvWt2LBhQ632LCsriyuvvDIGDhwYq1evrhL+JkkSp512WsyePTtj7wEAAAAAAACQCXkdAP/3f/93PPbYY9GuXbu0+fLTwI8++mj07t07Xn/99Rrt995770X//v1j9OjRsX379opAOeLT4Ldp06Zxyy23xKRJk6JTp04Zfx8AAAAAAACA+sjrADgi4pvf/Ga88cYbccwxx1Q5DZwkSSxZsiSOO+64uO2223a5z2OPPRa9e/eOmTNnVnvqd7/99ou//e1vMWLEiKy9CwAAAAAAAEB95H0AHBGx3377xdSpU+MnP/lJ2nz5Cd6tW7fGiBEj4swzz4w1a9akPbN169YYPnx4nHfeebFu3bpqw99zzjknZs+eHX369GmQ9wEAAAAAAACoi0YRAEdENG3aNG699dZ48skno2PHjtWeBn7qqaeiV69eMXXq1IiI+Pvf/x59+vSJu+++uyL43fHK5+bNm8cdd9wRjz76aJVrpgEAAAAAAAB2N40mAC53+umnx+zZs+O4446rNgT+5z//GSeddFIMGzYsjjnmmCguLq721O/BBx8c06dPj0suuSQXrwEAAAAAAABQa40uAI6I6NatW7z00kvxs5/9LC3YLT/hW1paGg888EBs2rSpYr5ckiRx0UUXxaxZs+LII49s8N4BAAAAAAAA6qpRBsAREQUFBfGrX/0qnn322ejcuXO1p4ErX/ncqlWruP/+++P++++PVq1a5ap1AAAAAAAAgDpptAFwua9+9atRXFwcPXr0SJvfMfhNkiQOP/zwmDVrVlx00UW5aBMAAAAAAACg3hp9APzRRx/F5ZdfHv/4xz8i4l+Bb/mJ4PIg+MMPP4xFixblrE8AAAAAAACA+mrUAfCcOXOid+/e8dBDD0VEpF37vOMJ4FQqFatXr46BAwfGlVdeGaWlpblsGwAAAAAAAKBOGm0AfMcdd0S/fv1i0aJF1X7vd8cTwOV/T5IkRo8eHf3794/33nsvV60DAAAAAAAA1EmjC4A3bNgQgwYNih//+MexZcuWiEj/3m9ExIgRI2L69OlxwAEHVDkVnCRJzJw5M3r16hWPPPJIzt4DAAAAAAAAoLYaVQA8Y8aM6NWrVzzxxBMVwW65JEmiY8eOMXHixLjllluiT58+MXv27BgyZEhFMBwRFUHwhg0bYsiQIfHv//7vFUEyAAAAAAAAwO6s0QTAv/vd72LAgAHx3nvvVRv+Hn/88fHmm2/G17/+9Yr51q1bx9ixY+Oee+6Jli1bpu1Xfhr4f/7nf6JPnz6xYMGCBnsXAAAAAAAAgLrI+wB4zZo1MXDgwPjpT38aW7durfKt31QqFT/72c/ixRdfjK5du1a7x/e///147bXX4tBDD61yGjhJknjrrbfi6KOPjvvuu69B3gkAAAAAAACgLvI6AJ46dWr06tUrJk2aVO2p3y5dusQzzzwTv/rVr6KgYNev2rNnz3j99ddj2LBh1V4J/fHHH8f3v//9+M53vhObNm3K2jsBAAAAAAAA1FVeB8AnnnhiLFu2rNrw96STToo333wzTj755Brv16JFi/jf//3fGDNmTLRu3Trtd+WngR966KE46qijYvbs2Rl7DwAAAAAAAIBMyOsAuKysLCIi7crngoKCuO6662Ly5Mmx55571mnfb3/72zFr1qzo1atXtVdCL1y4MPr371//FwAAAAAAAADIoLwOgHeUJEl07do1XnjhhRg5cmTaieC6+OIXvxjTp0+Pyy67rEoIHBHxySef1Gt/AAAAAAAAgExrFAFwkiRx6qmnxpw5c2LAgAEZ27ewsDB+//vfxxNPPBEdOnRIC4IBAAAAAAAAdjd5HwA3adIkbrrppnj66aejU6dOWalx1llnxezZs6Nfv35CYAAAAAAAAGC3ldcB8L777htTpkyJq666qkFr1fd6aQAAAAAAAIBsyOsAeM6cOXHsscc2WL3y08bPPPNMdOnSpcHqAgAAAAAAANREXgfAHTp0yEndr33tazFnzpyc1AYAAAAAAADYmbwOgHNpzz33zHULAAAAAAAAAGkEwAAAAAAAAACNhAAYAAAAAAAAoJEQAAMAAAAAAAA0Ek1z3UC+WLp0abz77rtpcwMGDMhRNwAAAAAAAABV7XYB8EknnZQ2/t73vhff+c53ctTNv/z5z3+OkSNHVoxTqVSUlpbmsCMAAAAAAACAdLtdAPzSSy9FKpWKJEkilUrFySefXKd9evfunTa+9NJLY9iwYfXqLUmSeq3/vFizZk1MmzYtFi9eHBs3bozmzZtH165d40tf+lIceuihuW6vinfffTdef/31eP/992Pz5s2xxx57xP777x99+/aNvffeO9ftAQAAAAAAQI3tdgFwpsyZMyctSP7www8zsm/5nruzkpKSeP3112PmzJkxY8aMmDlzZpX3HzVqVFx33XUZrTt16tT49a9/HZMnT46ysrJqn/niF78Yl112WVxyySVRWFiY0fq1kSRJPPjggzF69Oh48803q30mlUpF//7946c//WkMHDiwgTsEAAAAAACA2mu0AfDnzejRo2PmzJkxc+bMWLx4cYPW3rZtW1x++eVxxx13fOazixYtiv/8z/+M//3f/43HHnssDjzwwAboMN2KFSvivPPOi5dffnmXzyVJElOnTo2pU6fGoEGD4r777ovWrVs3UJcAAAAAAABQewLgRuKKK67ISd3S0tIYPHhwjB8/vlbr5s6dG/37948pU6Y06LXQH3zwQRx//PG1DskfffTRWLp0aTz//PNCYAAAAAAAAHZbBblugPx2zTXXVAl/O3XqFNdff33MmTMn1qxZEwsXLoy//OUv0adPn7TnVq1aFWeeeWZs2LChQXotLS2Nc889t0r4e9hhh8W9994b8+fPj7Vr10ZxcXGMHj069t1337TnXnvttfje977XIL0CAAAAAABAXQiAG6nWrVvHgAED4oorroiHHnooKzWKi4vjd7/7Xdpcz549Y86cOTFy5MgoKiqKDh06xBe/+MUYMmRITJ8+Pf7f//t/ac8vWrQofvnLX2alv8ruuuuumDZtWtrceeedF7Nnz45hw4bFIYccEu3bt48jjjgiLr/88iguLo4BAwakPT9u3LiYOHFig/QLAAAAAAAAtSUAbiSOOeaYGD58eNx3333x1ltvxfr162PKlClx6623xre+9a2s1Pz5z38eSZJUjNu0aRPPPPNMdOvWrdrnU6lU3HbbbXHWWWelzd95552xfPnyrPRY7uOPP44bb7wxbe7oo4+OsWPHRrNmzapd0759+5g4cWKVk8C/+MUv0t4bAAAAAAAAdhcC4EZixowZcccdd8TQoUOjZ8+eUVCQ3X+1c+bMiSeffDJt7pe//GXss88+n7n2rrvuipYtW1aMN2/eHL/97W8z3uOO7rnnnvjwww/T5v7nf/4nmjRpsst1bdq0iT/84Q9pc3PmzIkJEyZkvEcAAAAAAACoLwEwdTJu3Li08R577FHj7+Puvffece6556bNPfrooxnrrTqV+x0wYED06tWrRmvPPPPM6NGjR9rcI488krHeAAAAAAAAIFMEwNTJ+PHj08bnnHNOtGnTpsbrhw4dmjb+5z//GbNmzcpEa1WsWLEiXnvttbS57373uzVeX1BQEBdeeGHa3FNPPRWlpaUZ6Q8AAAAAAAAyRQBMrb3//vsxb968tLkTTjihVnv069cvCgsL0+aeeuqp+rZWrWeffTa2b9+eNlfbfis/v27dunj11Vfr2RkAAAAAAABklgCYWisuLq4y179//1rt0bJly+jdu3fa3Ny5c+vV185U7nevvfaKAw44oFZ79O3bt8r3grPVLwAAAAAAANSVAJhaW7BgQdq4adOmceCBB9Z6n0MPPXSX+2ZK5X0r162J1q1bx7777rvLfQEAAAAAACDXBMDU2vz589PG3bp1i4KCOe7QPAAAIABJREFU2v+nVDlQXbhwYZSVldWrt+pU7rd79+512kcADAAAAAAAwO5OAEytLV++PG1cORitqcpB7CeffBIlJSV17mtnstXvsmXL6twTAAAAAAAAZIMAmFrbtGlT2rht27Z12qe6dZX3rq+ysrLYsmXLZ9aticrrMt0rAAAAAAAA1FfTXDdA/qkcfLZs2bJO+1S3LtOhanX7ZarfTPW6cuXKWLVqVa3WLFq0KCO1AQAAAAAAaFwEwNTaRx99lDZu0aJFnfZpiAC4cq8Rmes3U73eeeedcf3112dkLwAAAAAAAD7fXAFNrSVJkjZOpVIZ2ac+ezVEjUy9NwAAAAAAAGSLAJhaa926ddp48+bNddqn8rd5IyL22GOPOu21M5V7jchcv5nuFQAAAAAAAOprt78CetKkSbF69eqc7zNjxox699BYZCoArm5ddYFtfWQyAK68LlO9Dh8+PAYPHlyrNYsWLYqzzz47I/UBAAAAAABoPHbrADhJkpg2bVpMmzatzuszsQ/pKgefGzZsqNM+1a3LdADcpEmTaNGiRdrp3Uz1m6leu3TpEl26dMnIXgAAAAAAAHy+7fZXQCdJUqefTO2zqz0/r7p27Zo2Xrp0aZ32qbyuefPm0bFjxzr3tTPZ6rfyvgAAAAAAAJBru30AnEql6vSTqX12tefn1SGHHJI2XrZsWWzfvr3W+7z//vtp4wMPPDCaNGlSr96qU7nfynVrqvK6Qw89tM49AQAAAAAAQDbstgFwfUPXbIS3QuBPVQ4+S0tLY+HChbXeZ8GCBWnjykFtplTud/78+bXe46OPPqpyAjhb/QIAAAAAAEBd7ZYBcCaua87mz+fdkUceWWXu1VdfrdUeW7ZsiTfeeCNt7ogjjqhXXztTud8PP/ww3n333VrtMX369CgrK0uby1a/AAAAAAAAUFdNc91AZbNnz851C3yG7t27x2GHHRbz5s2rmHvppZfi4osvrvEer776amzdujVt7owzzshYjzs69dRTo6CgIO2a6pdeein233//Gu8xZcqUtHH79u2jX79+GesRAAAAAAAAMmG3C4CLiopy3QI1cNZZZ6UFwI8//njccccd0bp16xqtf+CBB9LG++yzTxx11FEZ7bHcnnvuGX379o1p06al1a9pYJ0kSTz44INpc2eccUY0bbrb/e8DNBLbt2+PkpKSXLeRlzp27BgFBbvlBScAAAAAAA1CgkWdDB48OG666aaK8aZNm+JPf/pT/PjHP/7MtR9++GE88sgjaXODBg3KeI87Ou+889IC4ClTpkRxcXG111lX9uSTT8Z7772XNjd48OBMtwhQoaSkJLp06ZLrNvLSypUro3PnzrluAwAAAAAgZxyRoU6+9KUvxTe+8Y20uZEjR8ayZcs+c+1//Md/xObNmyvGLVq0iCuvvLJGdffbb79IpVIVP/vtt1+N1v3whz+MPffcM23uBz/4QZXv+la2adOmuOyyy9LmioqK4swzz6xRXQAAAAAAAGhIAmDq7Fe/+lWkUqmK8fr16+PrX/96LF++vNrnkySJESNGxOOPP542/x//8R/RtWvXrPbaqlWruPbaa9PmZsyYERdeeGGVbxGXW79+fQwcODDef//9tPnK7w0AAAAAAAC7C1dANxKzZs2qcq3yrjz//POxZcuWan/XoUOHuOqqqz5zj6KiovjJT34Sv/vd7yrm5s6dG7169Yof//jHceaZZ8a+++4ba9eujddffz1Gjx4dr732Wtoe//Zv/xYjR46scd/1cckll8TYsWNj+vTpFXN/+ctfori4OEaMGBH9+vWLLl26xLJly+L555+P0aNHVwl/Bw0aVOXkMwAAAAAAAOwuBMCNxNy5c+M3v/lNjZ9/5ZVX4pVXXqn2dz169KhRABwRcfPNN8c777wTTz75ZMXcqlWr4he/+EX84he/2OXaTp06xYQJE6Jt27Y17rs+mjZtGo899lgMGDAgFi9eXDH/9ttvx8UXX/yZ6/v06RN/+tOfstkiwE7tffW8KGjdKddt7Fa2b1odH9x8WK7bAAAAAADYrQiAqZfyUPU///M/46677qrxup49e8bjjz8eBx10UBa7q6pr164xderUGDx4cEydOrXG684555y4//77o02bNlnsDmDnClp3iiatO+e6DQAAAAAAdnO+AUy9FRYWxp133hkvv/xynHrqqVFQsPP/rA444IC4/fbbY/bs2Q0e/pbba6+94uWXX477778/jjzyyF0+279//5gwYUI89thjwl8AAAAAAAB2e04ANxJDhw6NoUOH5rSH448/Pp555pkoKSmJadOmxeLFi2Pjxo3RrFmz6Nq1a/Tu3TsOO6x+V3W+9957Gek1lUrFd7/73fjud78bS5YsiZkzZ8bSpUtjy5Yt0apVq9hvv/3iy1/+cnTt2jUj9QAAAAAAAKAhCIDJuI4dO8Y3vvGNXLdRYwcccEAccMABuW4DAAAAAAAA6s0V0AAAAAAAAACNhAAYAAAAAAAAoJEQAAMAAAAAAAA0EgJgAAAAAAAAgEZCAAwAAAAAAADQSAiAAQAAAAAAABoJATAAAAAAAABAIyEABgAAAAAAAGgkBMAAAAAAAAAAjYQAGAAAAAAAAKCREAADAAAAAAAANBICYAAAAAAAAIBGQgAMAAAAAAAA0EgIgAEAAAAAAAAaCQEwAAAAAAAAQCMhAAYAAAAAAABoJJrmuoGGsG3btnj77bdj1apVsXbt2li7dm188sknERHRr1+/OProo3PcIQAAAAAAAED9NdoA+J133on77rsv/va3v8Ubb7xREfhWdsMNN+wyAF69enUsWbIkbW6vvfaK7t27Z7RfAAAAAAAAgPpqdAHw3/72t7jpppviueeeiyRJIiIq/qwslUp95n4ff/xxHHfccVFWVlYxd9RRR8WMGTMy0zAAAAAAAABAhjSabwBv27YtRowYESeeeGI8++yzsX379kiSJJIkiVQqVeWnprp37x6DBw+u2CtJkpg1a1bMmzcvi28DAAAAAAAAUHuNIgBetWpV9O3bN2677baK4HdnYe/OTgPvyogRIyIi/cTwgw8+WL+mAQAAAAAAADIs7wPgjRs3xoknnhhz5sxJC34jouLEbkTEF77whTjwwAPrVONLX/pSHHzwwRHxaQicJEk89dRTmXkBAAAAAAAAgAzJ+wB46NChMW/evCrBbyqVinPOOSfGjx8fq1evjlWrVsWCBQvqXGfQoEFpp4fffvvtWLlyZb37BwAAAAAAAMiUvA6AJ0yYEE888UTa1cxJksThhx8eb775Zjz66KMxcODA6NChQ71rnXnmmVXmXnzxxXrvCwAAAAAAAJApeR0Ajxo1quLv5adzv/KVr8SMGTOiZ8+eGa1VVFQUhYWFaXNz587NaA0AAAAAAACA+sjbAPj111+PN998M+30b/fu3WP8+PHRokWLjNdr1qxZHHbYYWnXQNfnSmkAAAAAAACATMvbAHjixIkVfy//5u+NN94Ybdu2zVrNQw45JCIiUqlUJEkSCxcuzFotAAAAAAAAgNrK2wB42rRpaeN27drFt771razWrPwt4TVr1mS1HgAAAAAAAEBt5G0AvHDhwoqTuKlUKk488cQoKMju67Rv3z5tvHHjxqzWAwAAAAAAAKiNvA2AV61alTbu3r171ms2a9YsbfzRRx9lvSYAAAAAAABATeVtALx58+a0ceXTudmwdu3atHHlQBgAAAAAAAAgl/I2AG7ZsmXaeN26dVmvuXr16rRxmzZtsl4TAAAAAAAAoKbyNgDu0KFD2viDDz7Ies1Zs2ZFKpWqGO+zzz5ZrwkAAAAAAABQU3kbAO+///6RJEmkUqlIkiReffXVrNZbsWJFvPPOOxERFXUPPvjgrNYEAAAAAAAAqI28DYB79+6dNl62bFkUFxdnrd4DDzxQZa5Pnz5ZqwcAAAAAAABQW3kbAJ9wwglV5m655Zas1NqyZUvcfvvtadc/R0SceuqpWakHAAAAAAAAUBd5GwCfdtpp0aZNm4iIimugH3744XjhhRcyXuuyyy6LDz/8MCI+vf45IqJnz55xyCGHZLwWAAAAAAAAQF3lbQDcvHnzGDZsWEUgm0qloqysLM4///yYP39+xur89re/jXvvvTft9G8qlYpLL700YzUAAAAAAAAAMiFvA+CIiJ/+9KcVp4AjPg1mV61aFf369YsJEybUa+9NmzbFD37wg7j66qur/O6AAw6IYcOG1Wt/AAAAAAAAgEzL6wB47733jl//+tcVp4AjPg2B169fH9/85jfj5JNPjqeeeiq2bdtW4z2XLVsWv/nNb2L//fePP/3pT5EkScXp3yRJoqCgIO64445o2rRpxt8HAAAAAAAAoD7yPsW89NJLY+rUqTFu3LiKoLb8m8AvvvhivPjii9GmTZvo3bt3td/snT17dvzmN7+JJUuWxIwZM6K4uDgiIu1q6fJxKpWKK6+8Mk455ZQGejsAAAAAAACAmsv7ADgi4sEHH4xNmzbFpEmTqoTAEREbNmyIKVOmxJQpUyLiX+FukiTx+OOPx+OPP542X76+sgsuuCBuuummrL4LAAAAAAAAQF3l9RXQ5QoLC2P8+PExfPjwKiFu+U+SJGm/K1c+v+OJ3x3D3/LfXXHFFfHAAw9k/2UAAAAAAAAA6qhRBMAREU2aNIn//u//jokTJ0aPHj2qhL07hsE7m6/8uyRJYp999okJEybEb3/726y/AwAAAAAAAEB9NJoAuNzpp58ef//73+OPf/xjHHnkkVVO+EbsPAze8dkePXrErbfeGu+880584xvfaOjXAAAAAAAAAKi1RvEN4MoKCwvjhz/8Yfzwhz+M+fPnxzPPPBPTpk2L4uLieP/992PLli1pz6dSqdhrr73isMMOi759+8bpp58exx57bLXfAQYAAAAAAADYXTXKAHhHhx56aBx66KFx+eWXV8ytX78+Pv744ygrK4sWLVpEu3btorCwMIddAgAAAAAAANRfow+Aq9OuXbto165drtsAAAAAAAAAyKhG9w1gAAAAAAAAgM8rATAAAAAAAABAIyEABgAAAAAAAGgkBMAAAAAAAAAAjUTTXDdQXxs3bowkSSrGqVQq2rRpk3c1AAAAAAAAAOorr08Az5o1K9q3bx8dOnSo+LnwwgszXufCCy9Mq9GhQ4eYP39+xusAAAAAAAAA1EdeB8D33HNPJElS8RMRMXLkyIzXufbaa9PqJEkS99xzT8brAAAAAAAAANRH3gbAZWVlMW7cuEilUhU/xx13XPTu3TvjtY455pjo169fRHx6/XOSJDF27NiM1wEAAAAAAACoj7wNgF977bVYv359RETF6d8hQ4Zkrd7555+fNl69enW8/vrrWasHAAAAAAAAUFt5GwC/+OKLVebOOeecrNU799xzq8y98MILWasHAAAAAAAAUFt5GwDPnTs3bdy9e/fYc889s1Zvzz33jB49euyyBwAAAAAAAIBcytsAeN68eRXf402lUnHUUUdlveZRRx1VUS9JkliwYEHWawIAAAAAAADUVN4GwCtXrkwb77333lmvWbnGBx98kPWaAAAAAAAAADWVtwHwhg0b0sZf+MIXsl6zco2NGzdmvSYAAAAAAABATeVtALxt27a08SeffJL1mlu3bk0bb968Oes1AQAAAAAAAGoqbwPgPfbYI228atWqrNesfO108+bNs14TAAAAAAAAoKbyNgDu0qVL2njJkiVZr/nuu++mjRvi2mkAAAAAAACAmsrbAHi//faLJEkilUpFkiQxbdq02LRpU9bqbdq0KV555ZWKeqlUKvbbb7+s1QMAAAAAAACorbwNgI866qi08bZt22LChAlZqzd+/Pgq3x3u1atX1uoBAAAAAAAA1FbeBsAnnHBCxd/LT+WOHDmySkibCdu2bYtRo0ZFKpVKm//KV76S8VoAAAAAAAAAdZW3AfBJJ50U7dq1S5t7991349prr814rV/84hdVvjHcqlWrOP300zNeCwAAAAAAAKCu8jYALiwsjGHDhkWSJBHxr1PAv/vd7+LWW2/NWJ3Ro0fHLbfcUnH6t/z7vxdccEG0bNkyY3UAAAAAAAAA6itvA+CIiCuuuCJatWpVMS4Pga+66qr49re/HWvWrKnz3uvWrYvzzz8/rrzyyiq/a9GiRVx99dV13hsAAAAAAAAgG/I6AO7atWuMHDmy4hRw+encJEli3LhxcdBBB8VVV10VixcvrvGeS5YsiauuuioOPPDAePjhhyv23HH/a6+9Nnr06JGVdwIAAAAAAACoq6a5bqC+rrzyyvjrX/8azz33XEX4W/7nmjVr4tZbb41bb701unfvHn369ImePXtG+/bto3379pFKpWLdunWxdu3amDdvXsyYMSP+8Y9/RESkXS1dLpVKxde+9rX42c9+lpN3BQAAAAAAANiVvA+AU6lUPProo/HVr341Zs6cWRHY7nhqNyLiH//4R7z//vvx6KOP7nSv8md3XL/j74499th47LHHMv0KAAAAAAAAABmR11dAl2vdunX89a9/jTPOOCMtxI34NMgt/0mSZJc/Oz67oyRJ4uyzz47JkyfHHnvs0ZCvBgAAAAAAAFBjjSIAjojYY4894sknn4zf//730bp16ypBcER6GFzdT2VJkkTbtm3jrrvuiscffzxatWrVEK8CAAAAAAAAUCeNJgAud9lll8XixYvjyiuvjI4dO6ad8P0sOz7buXPn+NnPfhZLliyJH/3oRw3QOQAAAAAAAED95P03gKvTqVOn+M1vfhO/+tWv4oUXXohnnnkmZsyYEW+//XZs3Lix2jVt27aNww8/PPr27RunnXZanHjiidG0aaP8xwMAAAAAAAA0Uo064SwsLIzTTjstTjvttIq5DRs2xOrVq2Pz5s0REdGyZcvo1KlTtG3bNldtAgAAAAAAAGREow6Aq9O2bVthLwAAAAAAANAoNbpvAAMAAAAAAAB8XgmAAQAAAAAAABoJATAAAAAAAABAIyEABgAAAAAAAGgkBMAAAAAAAAAAjYQAGAAAAAAAAKCRaJrrBrJl27ZtMXfu3Hj77bdjzZo1sX79+ti4cWOUlZVlrMbo0aMzthcAAAAAAABAfTWqAHjjxo3x0EMPxQMPPBAzZ86M0tLSrNYTAAMAAAAAAAC7k0YTAP/xj3+Mq6++OjZu3BgREUmSZLVeKpXK6v4AAAAAAAAAtZX3AfCWLVvi7LPPjsmTJ6eFvtkMaLMdLgMAAAAAAADURV4HwGVlZTFo0KB47rnnImLnoa/AFgAAAAAAAPg8yOsA+M4774xJkyZVG/yWh74tWrSIgw46KDp06BBt27aNgoKChm6zUXrppZfixBNPzPi+L774YpxwwgkZ3zdT/V5xxRVx6623ZqAjAAAAAAAAyLy8DYA3bNgQ119/fZXwN0mSaN++ffzoRz+KIUOGxBFHHCH0zSNNmjTJdQsAAAAAAACQt/I2AJ40aVKsWbOmIgBOkiRSqVSceuqp8ec//zk6d+6c4w6prbZt28bRRx+d6zYAAAAAAAAgb+V1AFyuPPzt379/TJgwIQoLC3PY2edD9+7d46qrrqrz+nfffTfGjRuXNjdkyJBo2bJlfVurkbZt28Yll1xS63UDBgzIQjcAAAAAAACQGXkbAM+ZM6fK9c933XWX8LeBHHDAAXHzzTfXef0VV1xRZW7o0KH16Kh2OnToUK/+AQAAAAAAYHeUtx/HXbVqVdq4qKgoevbsmaNuqI3S0tIYM2ZM2tzBBx8cxx57bI46AgAAAAAAgMYhbwPgNWvWRMS/rn/u27dvjjuipp5++ulYsWJF2lxDnv4FAAAAAACAxipvA+DmzZunjbt06ZKjTqit+++/P21cUFAQF154YW6aAQAAAAAAgEYkbwPgvfbaK228efPmHHVCbZSUlMTEiRPT5k455ZTo1q1bjjoCAAAAAACAxiNvA+AjjjgikiSpGFe+Upjd09ixY2Pr1q1pc65/BgAAAAAAgMzI2wD4lFNOiYiIVCoVSZLEtGnTctwRNVH5+ucOHTrE2WefnZtmAAAAAAAAoJFpmusG6mrQoEHxk5/8JLZs2RIREYsXL4558+bFYYcdluPO2Jm33nor3njjjbS5IUOGVPmec0NZt25dvPrqq7F48eIoKSmJZs2axRe+8IXYe++949hjj/VdaQAAAAAAAPJO3gbAHTt2jB/96Edx++23RyqVioiIUaNGxSOPPJLjztiZ++67r8rcxRdfnINOIpYuXRodO3aM7du37/SZQw89NL73ve/Fj370o2jdunUDdgcAAAAAAAB1k7cBcETEL3/5y3j88cdj6dKlkSRJPP744/HQQw/FkCFDct0alZSWlsaYMWPS5g477LA45phjctLProLfcvPnz48RI0bEr3/967jjjjvi29/+dgN0BkBD2759e5SUlOS6jbzTsWPHKCjI26+JAAAAAECjldcBcOvWrWP8+PFx/PHHx0cffRRJksTQoUOjsLAwzj333Fy3xw6efvrpWLFiRdpcrk7/1tbatWvj/PPPj2nTpsUf/vCHjO+/cuXKWLVqVa3WLFq0KON9AHxelZSUuPa/DlauXBmdO3fOdRsAAAAAQCV5HQBHRBQVFcVzzz0XZ5xxRqxduza2bt0a5513XgwbNixGjRoV++yzT65bJCLuv//+tHGTJk3iggsuaPA+WrZsGSeddFKcdtppUVRUFAceeGC0a9cuUqlUlJSUxFtvvRXPP/983HvvvbF27dq0tf/1X/8VHTt2jFGjRmW0pzvvvDOuv/76jO4JAAAAAADA51NeB8AbNmyIiE+vEn7hhRfiggsuiHnz5kWSJPGnP/0pHnjggTjjjDPipJNOimOOOSa6dOkS7du3j6ZNM/Pabdu2zcg+jV1JSUlMnDgxbe7rX/967LXXXg3WQ6dOneK2226Liy++ONq1a1ftM926dYtu3brFqaeeGqNGjYorrrgi7rnnnrRnrrvuujjuuOPiq1/9akO0DQAAAAAAALWS1wFw+/btI5VKpc2Vj5MkidLS0pgwYUJMmDAh47VTqVSUlpZmfN/GaOzYsbF169a0uaFDhzZoD4cffngcfvjhNX6+devWcffdd0f37t3j5z//edrvrrnmmpgxY0amWwQAAAAAAIB6y+sAOOLToLc6OwbB5Fbl6587duwYAwcOzE0ztXTttdfG9OnT004wz5w5M15++eUYMGBARmoMHz48Bg8eXKs1ixYtirPPPjsj9QGoau+r50VB6065bmO3sX3T6vjg5sNy3QYAAAAAUAN5HwBXPgFcHvgmSRKpVKrK7zNBqFxzb731Vrzxxhtpc+eff340a9YsRx3V3o033ljlCutnnnkmYwFwly5dokuXLhnZC4DMKGjdKZq07pzrNgAAAAAAaq0g1w1kWnnom43gl9q77777qsw19PXP9XXEEUfEQQcdlDY3ZcqUHHUDAAAAAAAAO5f3AXCSJA3+Q82UlpbGmDFj0uaOPPLI6N27d446qrtjjz02bbxs2bIcdQIAAAAAAAA7l9dXQM+ePTvXLbALTz/9dKxYsSJtLt9O/5arfEXzqlWrctQJAAAAAAAA7FxeB8BFRUW5boFduP/++9PGhYWFccEFF+SmmXqqfPLbFeMAAAAAAADsjvL+Cmh2TyUlJTFx4sS0udNPPz06d+6co47qp/JJ5nx9DwAAAAAAABo3ATBZMXbs2Ni6dWva3MUXX5yjbupv2rRpaeO99947R50AAAAAAADAzgmAyYrK1z937tw5Tj/99Nw0U0+zZ8+ORYsWpc195StfyVE3AAAAAAAAsHMCYDLurbfeijfeeCNt7oILLojCwsIcdVQ/11xzTZW50047LQedAAAAAAAAwK4JgMm4++67r8rc0KFDM7J3KpVK+znhhBM+c82yZcvqXG/kyJHx7LPPps0dddRRMWDAgDrvCQAAAAAAANkiACajSktLY8yYMWlzvXv3jiOPPDJHHUVceuml8dWvfjWee+65KCsrq9GadevWxdChQ+OGG26o8rubb745UqlUptsEAAAAAACAemua6wZoXJ5++ulYsWJF2lymTv/WVZIk8de//jX++te/RufOnePMM8+MPn36RFFRUey1117Rrl27SKVSUVJSEm+99VZMnjw5Hnjggdi4cWOVva6//vo4+eSTc/AWAAAAAAAA8NkEwGTU/fffnzZu1qxZnH/++blpphqrVq2Ke++9N+69995arx0xYkSMHDkyC10BAAAAAABAZnwuAuDly5fH2rVrY926dbF+/fooLS3NyL4nn3xytGrVKiN7NQYlJSUxceLEtLmBAwdGx44dc9RRZnTr1i3uueeeOP3003PdCgAAAAAAAOxSowyAFy5cGGPGjInp06fHjBkzYv369VmpM3/+/DjooIOysnc++stf/hJbt25Nm8v19c8RETfccEP07ds3XnrppZg1a1aUlJR85pqWLVvGl7/85fj+978fgwcPjsLCwgboFAAAAAAAAOqnUQXAr7zyStxwww3x/PPPR5IkEREVf2ZaKpXKyr757NJLL41LL700qzXq8u/ziCOOiCOOOCKuueaaiIhYtmxZLFq0KJYuXRolJSXx0UcfRSqVinbt2kWHDh3ii1/8YvTq1UvoCwAAAAAAQN5pFAFwkiRx3XXXxY033hjbt29PCwmzEdRmK1SmYXTr1i26deuW6zYAAAAAAAAg4xpFADxkyJB49NFHK4LZnYW+lYPbXYXD1YW8Tv0CAAAAAAAAu7O8D4Cvv/76eOSRRyIiPaDdMcAtLCyMJk2axJYtWyKVSkWSJJFKpaJt27axdevW2Lx5c9qeqVSqYq/yZ1u1ahVNm6b/42rSpEm2XgsAAAAAAACg1gpy3UB9vP322/HLX/4yLbCN+DS0Pfnkk+Phhx+OlStXxieffBLXXnttlfVr166Njz76KLZs2RJLly6N8ePHx1VXXRX77rtv2mnFd0J2AAAgAElEQVTiJEmie/fu8cILL8TatWsrfv7t3/6twd4VAAAAAAAA4LPkdQB84403pp30TZIkmjZtGn/+85/jueeei8GDB0enTp0+c59mzZpFt27dYuDAgXHTTTfFkiVLYty4cbHPPvtUnABesGBBHH/88fHEE09k85UAAAAAAAAA6ixvA+APPvggxo0bV+Wq5vvuuy8uuOCCeu1dUFAQgwYNirlz58Y3v/nNipB58+bN8a1vfSuee+65evcPAAAAAAAAkGl5GwC/9NJLUVZWFhH/Cn+/8Y1vxPnnn5+xGm3bto1x48bFWWedVVGjtLQ0vv3tb8fKlSszVgcAAAAAAAAgE/I2AH755ZerzF1xxRUZr9OkSZMYM2ZM7LPPPhVz69ati2uuuSbjtQAAAAAAAADqI28D4Dlz5qSNu3TpEgMGDMhKrVatWlV8bziVSkWSJDFmzBingAEAAAAAAIDdSt4GwKtXr64IY1OpVBx99NG13mPz5s01fnbw4MGxxx57VIy3bdsW48ePr3VNAAAAAAAAgGzJ2wB4zZo1aeP9999/l883bdq0ylxtAuDmzZvHCSecEEmSVMy9+OKLNV4PAAAAAAAAkG15GwBv3Lgxbdy+fftdPt+mTZsqc+vXr69VzR49ekREVJw8njdvXq3WAwAAAAAAAGRT3gbArVq1ShunUqldPl9dALx06dJa1ezUqVPa+J///Get1gMAAAAAAABkU94GwG3btk0br1u3bpfPV3dC+B//+Eetan788cdp402bNtVqPQAAAAAAAEA25W0AvNdee6V9j3ft2rW7fP6ggw6qMjd9+vRa1Vy8eHHauLrvCgMAAAAAAADkSt4GwIceemhE/Ot7vAsWLNjl8wceeGC0bNkybc2LL75Y43rbtm2Ll19+Oe2q6Y4dO9ahcwAAAAAAAIDsyPsAuNzbb7+ddiK4soKCgigqKkp75u9//3tMmTKlRvUefPDBKCkpiYiIJEkilUpFjx496tA5AAAAAAAAQHbk7R3G/fr1Sxtv2bIliouLo6ioaKdrzj333LRrn5MkiR//+Mcxbdq0aPX/2bv3KKvLen/gn9kzXB0BHWYQwgAvGaboQU28gGRqJnhBGVQEYbQ07dAxtbD85a0WmpdMO8fEShgJDC+QZMfL0QQzJeN0AfGGVyCBgQlQbsYw+/dHizluhtsMe2bP/vJ6rcXS7zPf5/m+d45rVe/9PN/27bc5b8GCBXH11Vdn7P6NiBgwYEAj0wMA7H5qa2vrvlDHzispKYlUKm+/twkAAABAM8vbArhfv37Rrl272LBhQ93Y9OnTt1sAn3/++TF27Ni6HbzpdDpeeeWVGDhwYPziF7+IQw89tN6cadOmxeWXXx6rVq2qVwAPHjw4ex8IACDhqquro6ysLNcx8k5VVVWUlpbmOgYAAAAAeSJvtxK0bt06+vfvn1HmTps2bbtzunXrFiNGjMg4BjqdTsecOXPi8MMPj8MPPzxGjBgRY8aMieHDh0ePHj2ivLw8qqqq6srfzc87/vjjo1+/fk36GQEAAAAAAAAaIm93AEdEDBs2LJ5++um66/nz58dLL70UxxxzzDbn3HbbbTFjxoxYvXp1RqmbTqdj7ty5MW/evLp7NxfFW+78bdWqVdx+++3Z/CgAAAAAAAAAuyxvdwBHRJx99tnRunXruqI2nU7HzTffvN05paWlMWXKlGjdunXdWEFBQb0yePNO30+Wv5ufc9ddd8VRRx2V7Y8DAAAAAAAAsEvyegdwp06dYsaMGbFy5cq6sVRqx532qaeeGtOmTYsLLrgg492+W+70/aR0Oh1t2rSJ8ePHx4UXXrjr4QEAiK7XvBqp4s65jtFi1K5ZEUtuOTjXMQAAAADIY3ldAEdEnHLKKY2a9+UvfzkWLFgQ3/3ud2Py5Mmxbt26bd6bSqVi2LBhccMNN8RBBx3U2KgAAGwhVdw5CotLcx0DAAAAABIj7wvgXVFSUhLjx4+Pu+66K5599tmYM2dOLFu2LKqrq2OPPfaI0tLS6Nu3b5x00klRUlKS67gAAAAAAAAA27VbF8CbtW3bNgYNGhSDBg3KdRQAAAAAAACARtvxC3MBAAAAAAAAyAsKYAAAAAAAAICEUAADAAAAAAAAJIQCGAAAAAAAACAhFMAAAAAAAAAACaEABgAAAAAAAEgIBTAAAAAAAABAQhTlOsBmM2bM2ObPzjjjjAbPaQ7bygUAAAAAAACQCy2mAD7rrLOioKCg3nhBQUHU1NQ0aE5z2F4uAAAAAAAAgFxoMQXwZul0ulnmAAAAAAAAACRNiyuAP7mjd2eL3ebeBaxwBgAAAAAAAFqiVK4DAAAAAAAAAJAdLWYHcIcOHRq8k7cxcwAAAAAAAACSqsUUwKtWrWqWOQAAAAAAAABJ5QhoAAAAAAAAgIRQAAMAAAAAAAAkhAIYAAAAAAAAICEUwAAAAAAAAAAJUZTrAI31yCOPxKuvvpoxdtZZZ0WfPn1ylAgAAAAAAAAgt/K2AP7BD34Q8+bNq7tu06ZNXHHFFTlMBAAAAAAAAJBbeVsAv/vuuxERkU6nIyLi2GOPjQ4dOuQyEgAAAAAAAEBO5e07gNevX1/39wUFBdGzZ8/chQEAAAAAAABoAfK2AC4uLs647tatW46SAAAAAAAAALQMeVsAb1n4rlu3LkdJAAAAAAAAAFqGvC2Ae/fuHel0OgoKCiIiYunSpTlOBAAAAAAAAJBbeVsADxw4sO7v0+l0vPjii7kLAwAAAAAAANAC5G0BfOaZZ0ZhYWHd9fvvvx9z587NYSIAAAAAAACA3MrbArh79+4xZMiQjGOgv/vd7+Y4FQAAAAAAAEDu5G0BHBFx2223Rfv27SPiX8dAP/HEEzF+/PgcpwIAAAAAAADIjbwugHv06BE///nP63YBp9Pp+PrXvx7/+Z//metoAAAAAAAAAM0urwvgiIjzzjsvfvazn0VhYWEUFBREbW1t/Md//EeceOKJ8dJLL+U6HgAAAAAAAECzKcp1gF3x4YcfRkREeXl5dO3aNS699NL44IMPIp1Ox6xZs+L444+Pz3zmM3HaaadF3759o3fv3tGpU6fo2LFjtGrVapef36FDh11eAwAAAAAAACBb8roA7tSpUxQUFNQb33wcdETEG2+8EW+++WbWn11QUBA1NTVZXxcAAAAAAACgsfK6AI6IuqJ3S58shrd1DwAAAAAAAECS5H0BvOUO4M1l7+a/FhQUbHWX8K5QKAMAAAAAAAAtUd4XwFvKdtkLAAAAAAAAkC/yvgC2GxcAAAAAAADgX/K6AP7LX/6S6wgAAAAAAAAALUZeF8CHHXZYriMAAAAAAAAAtBipXAcAAAAAAAAAIDsUwAAAAAAAAAAJoQAGAAAAAAAASAgFMAAAAAAAAEBCKIABAAAAAAAAEkIBDAAAAAAAAJAQCmAAAAAAAACAhFAAAwAAAAAAACREUa4DNIcPPvggVq5cGatWrYrVq1dHTU1NVtY96aSTon379llZCwAAAAAAAGBXJbIAXrBgQUyePDlmz54dL7/8cqxevbpJnvPaa6/FZz7zmSZZGwAAAAAAAKChElUA/+EPf4jvf//78cwzz0Q6nY6IqPtrthUUFDTJugAAAAAAAACNlYgCOJ1Oxw033BDjxo2L2trajNK3KYrapiqVAQAAAAAAAHZFIgrg8847Lx555JG6YnZbpe+Wxe32yuGtlbx2/QIAAAAAAAAtWd4XwDfeeGM8/PDDEZFZ0H6ywG3VqlUUFhbGhg0boqCgINLpdBQUFESHDh3in//8Z6xfvz5jzYKCgrq1Nt/bvn37KCrK/I+rsLCwqT4WAAAAAAAAQIOlch1gV8yfPz9uuummjMI24l+l7UknnRRTp06Nqqqq+Pjjj+Paa6+tN3/lypWxdu3a2LBhQyxatCgee+yxGDt2bOy7774Zu4nT6XR8+tOfjmeffTZWrlxZ92f//fdvts8KAAAAAAAAsCN5XQCPGzcuY6dvOp2OoqKieOCBB+Lpp5+O8vLy6Ny58w7Xad26dXzqU5+K008/PW6++eZ455134qGHHoru3bvX7QB+/fXXo3///jF9+vSm/EgAAAAAAAAAjZa3BfCSJUvioYceqndU84QJE2LEiBG7tHYqlYqhQ4fGvHnzYsiQIXUl8/r16+Pcc8+Np59+epfzAwAAAAAAAGRb3hbAM2fOjE2bNkXE/5W/gwcPjuHDh2ftGR06dIiHHnoozjzzzLpn1NTUxPnnnx9VVVVZew4AAAAAAABANuRtAfz888/XG7vqqquy/pzCwsKYPHlydO/evW5s1apV8Z3vfCfrzwIAAAAAAADYFXlbAP/1r3/NuC4rK4sBAwY0ybPat29f977hgoKCSKfTMXnyZLuAAQAAAAAAgBYlbwvgFStW1JWxBQUFceSRRzZ4jfXr1+/0veXl5bHHHnvUXW/cuDEee+yxBj8TAAAAAAAAoKnkbQH8j3/8I+O6V69e272/qKio3lhDCuA2bdrEwIEDI51O140999xzOz0fAAAAAAAAoKnlbQH80UcfZVx36tRpu/fvueee9cZWr17doGf26NEjIqJu5/Grr77aoPkAAAAAAAAATan+ttg80b59+4wSuKCgYLv3b60AXrRo0Q53Dn9S586dM64XL16803Npmd59992YM2dOLFy4MNavXx977LFH9OrVK44++ujo2rVrruMBAAAAAABAg+RtAdyhQ4eMAnjVqlXbvX9rO4Tff//9Bj1z3bp1Gddr1qxp0Pyk2VHpvjOOOOKImDNnThbS7Lx0Oh2TJk2KH/3oR/G3v/1tq/cUFBTEcccdF9/+9rfj9NNPb9Z8AAAAAAAA0Fh5ewT0Pvvsk/E+3pUrV273/s985jP1xmbPnt2gZ7799tsZ11t7rzAt27Jly2LgwIExatSobZa/Ef8qiV944YU444wzory8fLcv+wEAAAAAAMgPeVsA9+7dOyL+7328r7/++nbvP/DAA6Ndu3YZc5577rmdft7GjRvj+eefz9j1WlJS0ojk5MqSJUviuOOOi+eff75B8x555JE46aSTlMAAAAAAAAC0eHm7hXVzAbzZ/PnzI51Ob/NY4lQqFYcddljMnj277p433ngjZs2aFSeccMIOnzdp0qSorq6uK48LCgqiR48eu/5BEuRrX/tadOzYsUFzunfv3kRpMtXU1MQ555xTbxf3wQcfHFdddVUce+yxsc8++8SiRYvimWeeiTvvvDMWLVpUd98f//jHuPjii2Pq1KnNkhcAAAAAAAAaI28L4GOPPTbjesOGDTF37tw47LDDtjnnnHPOyTj2OZ1Oxze+8Y146aWXon379tuct2DBgrj66qvrlcsDBgxoZPpkGjt2bPTs2TPXMbbqpz/9abz00ksZY8OGDYtJkyZF69at68Y6deoUhx56aFRUVMSZZ56ZsVv4oYceipEjR8bgwYObLTcAAAAAAAA0RN4eAd2vX7+6I503mz59+nbnnH/++ZFK/esjby5zX3nllRg4cGDMmzdvq3OmTZsW/fv3j1WrVtX7mSIwP6xbty7GjRuXMXbkkUfGlClTMsrfT+rUqVM8/vjjse+++2aMf+9738t49zQAAAAAAAC0JHlbALdu3Tr69+9fdxxzOp2OadOmbXdOt27dYsSIERkFXjqdjjlz5sThhx8ehx9+eIwYMSLGjBkTw4cPjx49ekR5eXlUVVXVFcabn3f88cdHv379mvQzkh333XdfLF26NGPsZz/7WRQWFm533p577hl33313xthf//rXmDFjRtYzAgAAAAAAQDbkbQEc8a8jfD9p/vz59Y753dJtt90WnTp1ioh/7QL+ZLE7d+7cePDBB+Oee+6JqVOnxqJFi7b6XuFWrVrF7bffnsVPQlN66KGHMq4HDBgQhx9++E7NPeOMM+q96/nhhx/OWjYAAAAAAADIprwugM8+++xo3bp13Y7edDodN99883bnlJaW1jv6d8siePOfT45v/llExF133RVHHXVUtj8OTWDZsmXxxz/+MWNs1KhROz0/lUrFyJEjM8Z++9vfRk1NTVbyAQAAAAAAQDYV5TrArujUqVPMmDEjVq5cWTe2+R2/23PqqafGtGnT4oILLohVq1bVlbxb7vT9pHQ6HW3atInx48fHhRdeuOvhaRZPPfVU1NbWZowNHDiwQWsMHDgwfvCDH9Rdr1q1Kl588cUYMGBANiICAAAAAABA1uR1ARwRccoppzRq3pe//OVYsGBBfPe7343JkyfHunXrtnlvKpWKYcOGxQ033BAHHXRQY6OSA3Pnzs243meffWK//fZr0BpHH310FBYWxqZNm+rG5s2bpwAGAAAAAACgxcn7AnhXlJSUxPjx4+Ouu+6KZ599NubMmRPLli2L6urq2GOPPaK0tDT69u0bJ510UpSUlOQ6bov38ccfxx/+8Id45ZVXYvny5bFp06YoKSmJzp07xxFHHBEHHnhgs2d6/fXXM6579+7d4DWKi4tj3333jffee2+b6wIAAAAAAEBLsFsXwJu1bds2Bg0aFIMGDcp1lLx2yCGHbPfduF27do3y8vK46qqr4tOf/nSzZHrttdcyrhv7XAUwAAAAAAAA+WDHL8yFnbS98jciYsmSJXH33XfHAQccENdcc80O78+GDz74ION63333bdQ6WxbHf//73xudCQAAAAAAAJqKAphmt3HjxvjhD38YJ554YqxevbrJnrNp06bYsGFDxliHDh0atdaW89asWdPoXAAAAAAAANBUWtwR0M8991x84QtfyHUMdlIqlYrPf/7zMWjQoDjyyCOjd+/eUVJSEm3atImVK1fGO++8E7NmzYpf/OIXsWDBgoy5v//972Po0KHxxBNPRFFR9n8Vt1bStmvXrlFrbTkvmwVwVVVVLF++vEFz3nrrraw9HwAAAAAAgORocQXwF7/4xejVq1dceOGFceGFF0avXr1yHYlt+Na3vhWXXXbZNv8ZlZWVRVlZWfTr1y++9a1vxb333htXXnllfPzxx3X3PPPMM3HTTTfFTTfdlPV8a9eurTfWtm3bRq3VlAXwPffcEzfeeGPW1gMAAAAAAGD31SKPgH7vvffipptuigMPPDC+8IUvxKRJk2LdunW5jsUWbr311p0u6FOpVFx++eXxxBNPRKtWrTJ+9qMf/Siqqqqyni+dTtcbKygoyMpajV0HAAAAAAAAmlKLLIAj/lW41dbWxvPPPx+jR4+OffbZJ77yla/ECy+8kOto7IIvfOELMW7cuIyxtWvXxr333pv1ZxUXF9cbW79+faPW2vJdwnvssUej1gEAAAAAAICm1OKOgN5s8w7LzTsv16xZExMmTIgJEybEfvvtFxUVFTFy5MjYd999cxmTRhgzZkzceeed8cEHH9SNPfnkk3Hddddl9TnZLIC3nLe1tRvr8ssvj/Ly8gbNeeutt+Kss87KWgYAAAAAAACSocUWwJt98qjdzWXw22+/Hd/73vfiuuuuixNPPDEqKipiyJAhjX6/K82rTZs2MXjw4Ljvvvvqxl5++eVYv359vXft7orCwsJo27Ztxu7dDz/8sFFrbTkvmwXw5nclAwAAAAAAwK5qcUdAn3LKKZFKpbb5/tbNfzYfEf3ss8/GiBEjomvXrvG1r30tZs+enYPUNNQxxxyTcb1p06ZYtmxZ1p/TrVu3jOtFixY1ap0t5225LgAAAAAAALQELa4AfvLJJ2PhwoVx8803x2c/+9lIp9PbLYM3/3z16tXxs5/9LI477rjo3bt33HrrrbFkyZIcfAJ2xtZ2vC5fvjzrz/nsZz+bcb1w4cJGrbPlvN69ezc6EwAAAAAAADSVFnkEdNeuXWPs2LExduzY+NOf/hQTJkyIqVOnxsqVKyMi81jorR0R/cYbb8R3vvOduPbaa+Pkk0+Oiy66KM4444xo3bp1834QtmlbpX629e7dO/77v/+77vq1115r8Bpr166ttwN4y2IZAKClqa2tjerq6lzHyDslJSWRSrW478kCAAAA7LQWWQB/0lFHHRVHHXVU/PjHP47HHnssKisr46mnnopNmzZFxPbL4E2bNsVTTz0VTz31VHTq1CmGDx8eo0aNiiOPPLLZPweZtnbcc2lpadaf06dPn4zrpUuXxrvvvhu9evXa6TVmz55d9/u22aGHHpqVfAAATaW6unqrp66wfVVVVU3y30sBAAAAmkvefLW9devWUV5eHo8//ngsXrw4br311jjkkEN2+ojolStXxj333BNHH310HHrooXHnnXdGVVVVDj4JEREvvfRSxnVhYWGT/B+UX/rSl+rt4Jg5c2aD1pg1a1bGdadOneLYY4/d1WgAAAAAAACQdXlTAH9Sly5d4uqrr465c+fGnDlz4utf/3rsvffeWy2DNxfBnyyD58+fH1dffXV07949zjzzzJg+fXrU1NTk6NPsftavXx+PP/54xthRRx0V7dq1y/qzunTpEkcffXTGWGVl5U7PT6fTMWnSpIyxQYMGRVFRi988DwAAAAAAwG4oLwvgT+rbt2/85Cc/iSVLlsSjjz4aZ5xxRhQVFW23DI74V7FXU1MTjz/+eAwdOjS6desW3/zmN+Nvf/tbLj7GbuXHP/5xLF26NGPs1FNPbbLnDRs2LON61qxZMXfu3J2a+5vf/Cbee++9jLHy8vJsRQMAAAAAAICsSsw2xqKiohgyZEgMGTIkVqxYEb/85S+jsrKyrtDd3ruCIyJWrFgRd999d9x9993Rp0+fuOiii2L48OFRUlLSvB8kDyxdujRKS0ujsLCwwXOffvrpuO666zLG2rdvH5deeukO5/bs2TPef//9uusePXrUK2e35pJLLolbbrkl473DX/3qV+PFF1/c7mdYs2ZNjBkzJmPssMMOizPOOGOHzwQAaIm6XvNqpIo75zpGi1G7ZkUsueXgXMcAAAAAyKrEFMCf1Llz57jiiiviiiuuiLlz58aECRPiwQcfrHvn747K4L/97W9xxRVXxLe+9a0YNGhQVFRUxGmnnVbvXbK7q1/96lfx05/+NK655pooLy+P4uLiHc6pqamJn/zkJzF27Nh6x21feeWVsc8++zRV3Gjfvn1ce+218Y1vfKNu7OWXX46RI0fGxIkTo3Xr1vXmrF69Os4666xYuHBhxvgPfvCDjN8ZAIB8kiruHIXFpbmOAQAAAEATSnyj2adPn7jzzjvj73//ezz22GMxZMiQaNWq1U4dEf3Pf/4zfv3rX8eZZ54Zn/rUp+Lb3/52zJ8/Pxcfo8V5880346KLLoqysrI4++yz44477ojf/e538fbbb8eKFStizZo1sWjRopg1a1bceOONccABB8SVV14ZGzduzFjnlFNOqbcjuClcdtll0a9fv4yxBx98MPr27RsTJ06MN998M1atWhXz58+Pu+66K/r06RMzZ87MuH/o0KExePDgJs8KAAAAAAAAjZXIHcBbU1hYGKeffnqcfvrp8Y9//CMmT54clZWV8ec//zkidrwreNmyZXHHHXfEHXfcEUcccURUVFTEZZdd1rwfogVav359TJ8+PaZPn97guSeeeGI88sgj0apVqyZIlqmoqCgeffTRGDBgQLz99tt14/Pnz4+Kioodzv/85z8f999/f1NGBAAAAAAAgF2W+B3AW7P33nvHmDFjYs6cOTFv3ry48soro0uXLtvdFVxQUFD38zlz5sS///u/5yh9/mvfvn388Ic/jP/5n/+JPffcs9me261bt3jhhRfi+OOPb9C8s88+O5555plmzQoAAAAAAACNsVsWwJ/0uc99Lm6//fZYvHhxPP7441FeXh6tW7fOKIM3//WTR0Tvzs4999y45557ory8PHr06LFTc1KpVBx22GFx6623xuLFi+Pb3/52Tt6pvM8++8Tzzz8fEydOjD59+mz33uOOOy5mzJgRjz76qPIXAAAAAACAvLDbHAG9I6lUKk477bQ47bTTYtWqVfHggw9GZWVlvPzyy3W7fyNCARwRXbt2jcsuu6zuCOxVq1bF66+/HosWLYqlS5fG2rVro6amJjp06BB77bVXdO/ePY488shdLlHfe++9LKT/1z/DUaNGxahRo+Kdd96JP/3pT7Fo0aLYsGFDtG/fPnr27Bn9+vWLbt26ZeV5AAAAAAAA0FwUwFux5557Rrdu3aJr165RWFgYtbW1GSUwmTp16hT9+vWLfv365TpKg+23336x33775ToGAAAAAAAAZIUC+BP+8pe/RGVlZUyZMiWqq6vrxhW/AAAAAAAAQD7Y7Qvgqqqq+OUvfxmVlZXxyiuvRMTWC19HPwMAAAAAAAAt3W5ZAG/cuDEee+yxqKysjKeeeio2bdqUUfpuq+zdfM/+++8fo0ePbo6oAAAAAAAAADtttyqAX3755aisrIxf/epXsWrVqoiIHRa/m39eXFwcQ4cOjYqKiujfv3/zBAYAAAAAAABogMQXwB988EFMmjQpKisr44033oiIHZe+m+8pKCiIAQMGREVFRZSXl0f79u2bJTMAAJA/amtro7q6Otcx8k5JSUmkUqlcxwAAAIDESWQBvGHDhpg+fXpMnDgxfve730VtbW2Djnj+9Kc/HaNGjYrRo0dHr169miUzAACQn6qrq6OsrCzXMfJOVVVVlJaW5joGAAAAJE6iCuA//OEPUVlZGQ8//HB8+OGHEfF/pe6OSt927drFkCFDoqKiIk488cRt3g8AAAAAAADQUuV9Abxw4cJ44IEHorKyMt55552I2PkjniMi+vXrFxUVFXHuuedGhw4dmj4wAAAAAAAAQBPJywJ43bp18cgjj8TEiRPj+eefj3Q63aDSt1u3bjFy5MgYPXp0HHTQQc2SGQAAAAAAAKCp5VUBPHPmzJg4cWJMmzYt1q5dGxE7f8RzmzZt4vTTT4+Kior40pe+FKlUqnlCAwAAu52u17waqeLOuY7RYtSuWRFLbjl419eprY3q6uosJNr9lJSU+N/BACOOTjgAACAASURBVAAAu4kWXwC//fbbUVlZGZMmTYqFCxdGRMOOeO7bt29UVFTE8OHDY6+99mr6wAAAwG4vVdw5CotLcx0jcaqrq6OsrCzXMfJSVVVVlJb6nQQAANgdtMgC+KOPPoqpU6dGZWVlvPjiixHRsNK3tLQ0RowYERUVFXHIIYc0fWAAAAAAAACAFqDFFcAXXHBB/PrXv44NGzZExI6PeN58T1FRUZx22mlRUVERgwYNiqKiFvfRAAAAAAAAAJpUi2tJH3zwwYzrHe32PeSQQ6KioiJGjBjhOCsAAAAAAABgt9biCuCIHZe+e++9d5x//vkxevToOOKII5ozGgAAAC1I12tejVRx51zHaFFq16yIJbccnOsYAAAA5EiLLIC3lE6no7CwME4++eSoqKiIM888M1q3bp3rWAAAAORYqrhzFBY7DQoAAAA2a7EF8ObdvgcddFCMHj06LrzwwujatWuOUwEAAAAAAAC0XC2yAN5zzz3j3HPPjdGjR8cxxxyT6zgAAAAAAAAAeaHFFcCTJk2Kc845J9q2bZvrKAAAAAAAAAB5pcUVwBdccEGuIwAAAAAAAADkpVSuAwAAAAAAAACQHQpgAAAAAAAAgIRQAAMAAAAAAAAkhAIYAAAAAAAAICEUwAAAAAAAAAAJoQAGAAAAAAAASIiiXAcAAAAAWp7a2tqorq7OdYy8U1JSEqmU79sDAAC5owAGAAAA6qmuro6ysrJcx8g7VVVVUVpamusYAADAbsxXUgEAAAAAAAASQgEMAAAAAAAAkBAKYAAAAAAAAICE8A5gAAAAYKd0vebVSBV3znWMFqN2zYpYcsvBuY4BAACQQQEMAAAA7JRUcecoLC7NdQwAAAC2wxHQAAAAAAAAAAmhAAYAAAAAAABICAUwAAAAAAAAQEIogAEAAAAAAAASoijXAQAAAAB2Z7W1tVFdXZ3rGHmnpKQkUil7GwAAYEsKYAAAAIAcqq6ujrKyslzHyDtVVVVRWlqa6xgAANDi+JokAAAAAAAAQEIogAEAAAAAAAASQgEMAAAAAAAAkBDeAQwAAADQwnS95tVIFXfOdYwWo3bNilhyy8HZWau2Nqqrq7Oy1u6kpKQkUil7SQAA8oECGAAAAKCFSRV3jsLi0lzHSKTq6uooKyvLdYy8U1VVFaWlficBAPKBr+0BAAAAAAAAJIQCGAAAAAAAACAhFMAAAAAAAAAACeEdwAAAAADs1rpe82qkijvnOkaLUbtmRSy55eBcxwAAoJEUwAAAAADs1lLFnaOwuDTXMQAAICsUwAAAAABA1tXW1kZ1dXWuY+SdkpKSSKW8uQ8AaDwFMAAAAACQddXV1VFWVpbrGHmnqqoqSkvtSAcAGs9XyQAAAAAAAAASQgEMAAAAAAAAkBAKYAAAAAAAAICE8A5gAAAAAKBZdL3m1UgVd851jBajds2KWHLLwbmOAQAkjAIYAAAAAGgWqeLOUVhcmusYAACJpgAGAAAAAMhjtbW1UV1dnesYeamkpCRSKW9KBCBZFMAAAAAAAHmsuro6ysrKch0jL1VVVUVpqV3pACSLrzYBAAAAAAAAJIQCGAAAAAAAACAhFMAAAAAAAAAACeEdwAAAAAAACdP1mlcjVdw51zFalNo1K2LJLQfnOgYANDkFMAAAAABAwqSKO0dhcWmuYyRSbW1tVFdX5zpG3ikpKYlUyqGkAM1BAQwAAAAAADupuro6ysrKch0j71RVVUVpqS8lADQHX7cBAAAAAAAASAgFMAAAAAAAAEBCKIABAAAAAAAAEsI7gAEAAAAAYBd0vebVSBV3znWMFqN2zYpYcsvBu75ObW1UV1dnIdHupaSkJFIp+/9gd6YABgAAAACAXZAq7hyFxaW5jpE41dXVUVZWlusYeaeqqipKS/0+wu7MV0AAAAAAAAAAEkIBDAAAAAAAAJAQCmAAAAAAAACAhPAOYAAAAAAAIC90vebVSBV3znWMFqN2zYpYcsvBuY4BtDAKYAAAAAAAIC+kijtHYXFprmMAtGiOgAYAAAAAAABICAUwAAAAAAAAQEIogAEAAAAAAAASQgEMAAAAAAAAkBAKYAAAAAAAAICEUAADAAAAAAAAJIQCGAAAAAAAACAhFMAAAAAAAAAACaEABgAAAAAAAEgIBTAAAAAAAABAQiiAAQAAAAAAABJCAQwAAAAAAACQEApgAAAAAAAAgIRQAAMAAAAAAAAkRFGuA5AcixcvjldeeSUWLlwYK1eujIiIvfbaK7p06RJHHXVUdOvWLccJAQAAAAAAINkUwDTasmXLYsaMGfHss8/Gc889F1VVVdu9f7/99ouLLrooLr300ujcuXMzpYx47733olevXru8zjnnnBOPPPJIFhIBAAAAAABA03AENA02Z86cOPHEE6Nbt25xySWXxNSpU3dY/kZEvPPOO/H//t//i/322y/uv//+ZkgKAAAAAAAAuxcFMA02Z86ceO6556K2trZR8z/66KO4+OKL47LLLstyMgAAAAAAANi9OQKarDjggAPi5JNPjoEDB0bv3r2jrKws2rZtG0uXLo0XX3wx7rvvvpg9e3bGnHvvvTfKysrixhtvbPa8Y8eObfCcQw89tAmSAAAAAAAAQPYogGm0oqKiOO+88+IrX/lKnHDCCVu9p2PHjnHQQQdFRUVFjB8/PsaMGRMbN26s+/m4ceNi2LBh8bnPfa65YkdExC233NKszwMAAAAAAIDm4AhoGiyVSsX5558fr732WkyaNGmb5e+WLr300hg/fnzGWE1NTdx0001NERMAAAAAAAB2OwpgGuziiy+OKVOmxAEHHNDguRUVFfUK4yeeeCL++c9/ZiseAAAAAAAA7LYUwDRYYWHhLs0fNWpUxvVHH30Uc+fO3aU1AQAAAAAAAAUwOXDYYYfVG1uyZEkOkgAAAAAAAECyKIBpdu3atas3tnbt2hwkAQAAAAAAgGRRANPsFi5cWG+srKwsB0kAAAAAAAAgWYpyHYDdz8yZM+uN7b///s2aYd26dTF79ux4/fXXY8WKFVFQUBAlJSVRVlYWRx99dOy7777NmgcAAAAAAACyQQFMs9q0aVNMnjw5Y6x3797Ro0ePZs3RsWPHqKmp2ebPe/XqFSNGjIhvfOMb0blz52ZMBgAAAAAAAI3nCGia1c9//vNYtGhRxti5557b7Dm2V/5GRLz77rvx/e9/P3r06BF33nlnM6UCAAAAAACAXaMAptksXrw4xo4dmzG21157xZgxY3KUaMfWrVsXV155ZQwdOjQ2btyY6zgAAAAAAACwXY6AplnU1NTE8OHDY/Xq1Rnj48aNi7333rtZMrRq1Sr69+8fp512Wvzbv/1bHHTQQdGpU6do1apVVFdXx5tvvhm/+93v4uc//3l88MEHGXMfffTRuOSSS2LChAlZz1VVVRXLly9v0Jy33nor6zkAAAAAAADIfwpgmsUVV1wRv//97zPGvvSlL8Wll17a5M9u165d3HTTTXHJJZdEly5dtnpP165do2vXrnHCCSfEtddeG+PGjYvvf//7UVtbW3fPxIkTo3///nHRRRdlNd8999wTN954Y1bXBAAAAAAAYPfkCGia3E9+8pP4r//6r4yx7t27x6RJk6KgoKDJn9+lS5f43ve+t83yd0utW7eOG264IR544IF6P7v++uvj448/znZEAAAAAAAAyAoFME1q6tSpccUVV2SMdezYMX77299GaWlpjlLtnAsuuCC+/vWvZ4wtXrw4pk6dmqNEAAAAAAAAsH2OgKbJPPnkkzFy5MiMY5TbtWsXv/nNb6JPnz45TLbzrrvuuvjFL34RGzZsqBt78skn48ILL8zaMy6//PIoLy9v0Jy33norzjrrrKxlAAAAAAAAIBkUwDSJF154Ic4555zYuHFj3VirVq3i4Ycfjv79++cwWcOUlZXFgAED4umnn64bmzVrVtafUVZWltU1AQAAAAAA2D05Apqs+/Of/xyDBw+OdevW1Y2lUql44IEHYtCgQTlM1jjHHHNMxvXSpUszdjUDAAAAAABAS6EAJqvmz58fp5xySqxevTpj/N57743zzjsvR6l2zZa7c2tra6O6ujpHaQAAAAAAAGDbFMBkzdtvvx0nn3xyvXL09ttvj69+9as5SrXr0ul0vbGCgoIcJAEAAAAAAIDtUwCTFYsWLYovfvGLsWTJkozxG264Ia666qocpcqOZcuWZVynUqnYe++9c5QGAAAAAAAAtk0BzC5btmxZfPGLX4z3338/Y/zKK6+M66+/Pkepsuell17KuO7SpUukUv7VAQAAAAAAoOXRYrFLVq5cGSeffHIsWLAgY/zSSy+NO+64I0epsmfp0qXx+9//PmPshBNOyFEaAAAAAAAA2D4FMI22Zs2aOPXUU2PevHkZ4yNGjIh77rknR6my6/rrr4+PP/44Y+zUU0/NURoAAAAAAADYPgUwjbJhw4Y4/fTT4+WXX84YP/vss2PixIlNckRyz549o6CgoO5Pz549dzjn73//e6Ofd//998d9992XMfapT30qhg0b1ug1AQAAAAAAoCkpgGmwmpqaKC8vj5kzZ2aMf/nLX44HH3wwCgsLcxNsK2677bY48sgj49FHH623k3db1q9fH9dcc0185StfqfezG2+8Mdq1a5ftmAAAAAAAAJAVRbkOQP65/vrr4/HHH88YKyoqigMPPDCuu+66Rq15xBFHRHl5eTbi1fO///u/MXTo0OjQoUMMHjw4+vXrF4cffnh07949OnbsGK1atYp//OMf8frrr8dzzz0X999/fyxfvrzeOhUVFXHxxRc3SUYAAAAAAADIBgUwDba1Y5Vramri7rvvbvSao0aNarICeLMPP/wwpkyZElOmTGnw3PPPP7/ecdAAAAAAAADQ0jgCGrZjr732igkTJsSUKVOiqMj3JQAAAAAAAGjZNFok2pgxY6JHjx4xc+bM+NOf/hRLlizZ4ZxWrVpF3759o6KiIkaOHBnt27dvhqQAAAAAAACw6xTANNjEiRNj4sSJzf7c9957r8Fz9t9///jmN78Z3/zmNyMiYvny5fHmm2/GwoULY/ny5bF27dqora2Njh07xl577RU9e/aMI444Itq2bZvl9AAAAAAAAND0FMDsVkpLS6O0tDSOO+64XEcBAAAAAACArPMOYAAAAAAAAICEUAADAAAAAAAAJIQCGAAAAAAAACAhFMAAAAAAAAAACaEABgAAAAAAAEgIBTAAAAAAAABAQiiAAQAAAAAAABJCAQwAAAAAAACQEApgAAAAAAAAgIRQAAMAAAAAAAAkhAIYAAAAAAAAICEUwAAAAAAAAAAJoQAGAAAAAAAASAgFMAAAAAAAAEBCKIABAAAAAAAAEkIBDAAAAAAAAJAQCmAAAAAAAACAhFAAAwAAAAAAACSEAhgAAAAAAAAgIRTAAAAAAAAAAAmhAAYAAAAAAABICAUwAAAAAAAAQEIogAEAAAAAAAASQgEMAAAAAAAAkBAKYAAAAAAAAICEUAADAAAAAAAAJIQCGAAAAAAAACAhFMAAAAAAAAAACaEABgAAAAAAAEgIBTAAAAAAAABAQiiAAQAAAAAAABJCAQwAAAAAAACQEApgAAAAAAAAgIRQAAMAAAAAAAAkhAIYAAAAAAAAICEUwAAAAAAAAAAJoQAGAAAAAAAASAgFMAAAAAAAAEBCKIABAAAAAAAAEkIBDAAAAAAAAJAQCmAAAAAAAACAhFAAAwAAAAAAACSEAhgAAAAAAAAgIRTAAAAAAAAAAAmhAAYAAAAAAABICAUwAAAAAAAAQEIogAEAAAAAAAASQgEMAAAAAAAAkBAKYAAAAAAAAICEUAADAAAAAAAAJIQCGAAAAAAAACAhFMAAAAAAAAAACaEABgAAAAAAAEgIBTAAAAAAAABAQiiAAQAAAAAAABJCAQwAAAAAAACQEApgAAAAAAAAgIRQAAMAAAAAAAAkhAIYAAAAAAAAICEUwAAAAAAAAAAJoQAGAAAAAAAASAgFMAAAAAAAAEBCKIABAAAAAAAAEkIBDAAAAAAAAJAQCmAAAAAAAACAhFAAAwAAAAAAACSEAhgAAAAAAAAgIRTAAAAAAAAAAAmhAAYAAAAAAABICAUwAAAAAAAAQEIogAEAAAAAAAASQgEMAAAAAAAAkBAKYAAAAAAAAICEUAADAAAAAAAAJIQCGAAAAAAAACAhFMAAAAAAAAAACaEABgAAAAAAAEgIBTAAAAAAAABAQiiAAQAAAAAAABJCAQwAAAAAAACQEApgAAAAAAAAgIRQAAMAAAAAAAAkhAIYAAAAAAAAICEUwAAA/H/27jw+puv/4/g7CSHWxBZ7BVWhYqdUhYqWItWi7VcpaqulG6pflFS/VLVoaeur1Fa6IbS6alNibS1f+74lgsSWWLLIPr8/POTnZoKZZDKTTF7PxyOPR8+Ze879zDG9M/d+7j0HAAAAAAAAgJMgAQwAAAAAAAAAAAAAToIEMAAAAAAAAAAAAAA4CRLAAAAAAAAAAAAAAOAkSAADAAAAAAAAAAAAgJMgAQwAAAAAAAAAAAAATqKQowOA84mKitL27dsVFham+Ph4eXh4qHr16mrWrJl8fHwcHZ6ZI0eOaPfu3YqKilJSUpJKliypWrVqqVWrVipTpoyjwwMAAAAAAAAAAAAsRgIYNvPTTz9p+vTp2rZtm0wmU5bbNGzYUKNGjVLfvn3l4uJi5wj/X0pKiv773/9qzpw5OnXqVJbbuLm56YknntD48ePVpk0bO0cIAAAAAAAAAAAAWI8poJFjcXFx6tmzpwIDA7V169a7Jn8lad++ferXr5/at2+vixcv2jHK/3fixAk1adJEr7/++l2Tv5KUlpam3377TY899pheffVVpaSk2DFKAAAAAAAAAAAAwHokgJEjcXFxCggIUHBwsFXtNm7cqDZt2ujChQu5FFnWjhw5otatW+vgwYNWtfvss8/Uq1cvpaam5lJkAAAAAAAAAAAAQM6RAEaOvPzyy9q+fbuhrnr16vrkk0904MABXb16VYcPH9aCBQvk6+tr2O7kyZPq0aOH3ZKqN27cULdu3XTlyhVDfcuWLfXdd9/pxIkTio6O1p49exQUFKSyZcsatvvxxx81fvx4u8QKAAAAAAAAAAAAZAcJYGTbTz/9pJUrVxrq/P39tX//fr3++ut6+OGH5enpKV9fXw0aNEh79uxRr169DNtv27ZN8+bNs0u8kydPNpvy+c0339Tff/+t559/XrVr11aZMmXUqFEjvfvuu9q7d6/q169v2H7GjBnav3+/XeIFAAAAAAAAAAAArEUCGNliMpk0ceJEQ1316tX1888/q3Tp0lm2KVKkiL799ls1bdrUUD916lQlJCTkWqySFBkZqblz5xrqunfvrlmzZsnFxSXLNlWrVtVvv/2mkiVLZtRl9b4BAAAAAAAAAACAvIIEMLLlxx9/1L59+wx1s2fPVokSJe7Zzs3NTQsWLDDUXbhwwazO1j788EMlJiZmlD08PMwSwlmpVq2a3nvvPUPd2rVrzd47AAAAAAAAAAAAkBeQAEa2rFixwlCuUaOGAgMDLWrbuHFjtWnTxlCXeSppWzKZTFq1apWhrlevXqpUqZJF7QcOHKjixYsb6nIzXgAAAAAAAAAAACC7SADDaikpKfrtt98MdX379pWrq+Ufp/79+xvKf//9ty5dumSL8Mzs2rVL58+fN9T169fP4vYlS5bUM888Y6j78ccfbRIbAAAAAAAAAAAAYEskgGG1v//+W9euXTPUtWvXzqo+Mm+fnp6u33//PYeRZS1zstrd3V2tW7e2qo/M8R48eFARERE5DQ0AAAAAAAAAAACwKRLAsNr+/fsNZTc3N7Vs2dKqPmrVqiVvb29D3YEDB3IcW1Yyx9ukSRMVLVrUqj4effRRs7rcihcAAAAAAAAAAADILhLAsNrRo0cN5erVq5utkWsJX1/fe/ZrK5n7zbxfSzz44IMqVKjQPfsFAAAAAAAAAAAAHI0EMKx25MgRQ7l69erZ6qdatWqGcm4kVNPS0nTixAlDXXbidXNzU6VKlQx1JIABAAAAAAAAAACQ15AAhtUiIyMN5cyJXEtlTsSeP38+2zHdzZUrV5ScnGyoy8vxAgAAAAAAAAAAADlR6P6bAEZxcXGGcqlSpbLVT+Z2N2/eVHp6ulxdbXdfQuZYs9qvpTK3y6pv5A3pcVccHUKekpvjwVgbMdb2xXjbD2NtP7k9Hoy3EZ9t+2Gs7Yvxth/G2n74jrQvPtv2w1jbF+NtP4y1/TAeALJCAhhWy5z49PDwyFY/WbWLi4vLdoI2K1klaW0Vr60SwJcuXdLly5etanP48GFD+eTJkzaJJT+KiYkxq4v6oJ4DIslfjh49qkuXLlnVhrHOHsbavhhv+2Gs7Sc7Yy0x3tnFZ9t+GGv7Yrzth7G2H74j7YvPtv0w1vbFeNsPY20/2f2OdBaZr5cnJSU5KBLAcUgAw2rx8fGGctGiRbPVjz0SwJljlWwXr60SwHPnztXkyZNz1Ef37t1tEgsKjrZt2zo6hAKDsbYvxtt+GGv7Yazti/G2H8bavhhv+2Gs7Yexti/G234Ya/tivO2HsbYfxtro7NmzatKkiaPDAOyKNYCRYy4uLtlqZzKZbNaXPfaRuS9bxwoAAAAAAAAAAADkFAlgWK148eKG8s2bN7PVT2Ji4n37zqkSJUqY1dkqXlvHCgAAAAAAAAAAAOQUU0DDaiVKlNC1a9cyytlNqGbVLquEbU7YMgGcuZ2tYh0+fLh69eplVZsbN25o165dKlWqlDw9PVWtWjUVKVLEJvEg506ePGmYlvuHH35Q7dq1HRiRc2O87Yexth/G2r4Yb/thrO2L8bYfxtp+GGv7Yrzth7G2L8bbfhhr+2Gs7YvxztuSkpJ09uzZjLK/v78DowEcgwQwrJY58Xnjxo1s9ZO5nYeHh1xdbftQelZJWlvFa6sEcIUKFVShQgWr27Vq1com+0fuq127turXr+/oMAoMxtt+GGv7Yazti/G2H8bavhhv+2Gs7Yexti/G234Ya/tivO2HsbYfxtq+GO+8hzV/UdAxBTSsVrlyZUP5zjtprJG5XeZ+baFcuXJyd3e/534tZY94AQAAAAAAAAAAgJwgAQyr1a1b11COiIjIVj+Z2/n6+mY7prtxc3Mzm3ojO/GmpaUpMjLSUJcb8QIAAAAAAAAAAAA5QQIYVsuc+IyIiFB8fLzV/Rw9etRQzpxYtpXM8R45csTqPk6ePKnU1FRDXW7FCwAAAAAAAAAAAGQXCWBYzc/Pz1BOS0vTjh07rOrj1KlTunDhgqGuQYMGOY4tK5nj3b17txITE63qY8uWLWZ1uRUvAAAAAAAAAAAAkF0kgGG1Vq1aydPT01AXGhpqVR8bN240lF1dXdWpU6echpalzp07G8rJycn6+++/reojc7wPP/ywqlevnuPYAAAAAAAAAAAAAFsiAQyrFS5c2CypumzZMplMJov7WLp0qaHcqlUrVahQwSbxZdasWTNVqVLlnvu/l9jYWK1Zs8ZQ9/TTT9skNgAAAAAAAAAAAMCWSAAjW5577jlDOSwsTD/99JNFbfft26dNmzYZ6nr16mWz2DJzcXFRz549DXUrV640m4L6bhYvXqy4uDhDXeb+AAAAAAAAAAAAgLyABDCy5emnn1bDhg0Nda+99ppZojSztLQ0DR482FBXsWJFs7q7cXFxMfy1a9fOonZvvfWWihYtmlFOSEjQyJEj79vu3LlzmjhxoqEuMDBQjRo1smi/AAAAAAAAAAAAgD2RAEa2uLi46D//+Y+h7syZMwoMDNSNGzeybJOcnKw+ffpo586dhvoJEyaoWLFiuRarJFWpUkXDhw831AUHB+utt96669TV58+f11NPPWV4P1m9bwAAAAAAAAAAACCvIAGMbOvWrZt69OhhqNuwYYP8/Pz06aef6vDhw7p27ZqOHTumRYsWqXHjxvruu+8M27dq1UqvvPKKXeINCgpSzZo1DXUzZsxQ69attXLlSp0+fVpXr17Vvn379N5776lRo0Y6cOCAYfvRo0fLz8/PLvECAAAAAAAAAAAA1irk6ACQvy1evFhnz57Vjh07MurOnDmj11577b5ta9WqpeDgYBUqZJ+PYalSpbR27Vr5+/srOjo6o/6ff/4xW9M4K4GBgZo2bVpuhggAAAAAAAAAAADkCAlg5EjJkiUVEhKifv36ac2aNRa3e+yxx7RixQpVrFgxF6MzV79+fW3dulU9evTQoUOHLG43fPhwffLJJ3ZLViN/K1++vIKCggxl5B7G234Ya/thrO2L8bYfxtq+GG/7Yazth7G2L8bbfhhr+2K87Yexth/G2r4YbwB5nYvpbgugAlZau3atpk+frm3btt11Gz8/P40aNUovvfSSXFxcrN5H5jb+/v4KDQ21up+UlBTNnTtXc+bM0enTp7PcxtXVVU888YTGjx+vxx57zOp9AAAAAAAAAAAAAPZGAhg2FxkZqX/++Ufh4eGKj4+Xh4eHqlWrpubNm5utwZsXHDp0SHv27FFkZKSSk5NVsmRJ1apVS61atVLZsmUdHR4AAAAAAAAAAABgMRLAAAAAAAAAAAAAAOAkXB0dAAAAAAAAAAAAAADANkgAAwAAAAAAAAAAAICTIAEMAAAAAAAAAAAAAE6CBDAAAAAAAAAAAAAAOAkSwAAAAAAAAAAAAADgJEgAAwAAAAAAAAAAAICTIAEMAAAAAAAAAAAAAE6CBDAAAAAAAAAAAAAAOAkSwAAAAAAAAAAAAADgJEgAAwAAAAAAAAAAAICTKOToAAAAQN6UnJyso0ePKjw8XOfPn1dsbKxSUlJUqlQplS1bVn5+fvL19ZWbm5ujQwWQz5w4cUJ79+7V2bNnlZCQIA8PD3l7e6tOnTry8/NT0aJFHR0iYLGkpCTt2bNHR44c0dWrV3Xz5k2VKlVKFSpUUJMmTVS7ohAT/QAAIABJREFUdm25uLg4OkynEBcXpy1btujs2bO6cuWKChUqpMqVK6tp06aqW7euo8Mr8BITE7Vt2zYdPXpUV69elbu7u6pWraqWLVuqZs2ajg4PQB524cIFHTt2TBEREbpy5YoSEhLk7u6u0qVLq0aNGmrWrJnKlCnj6DABAMhXSAADAPINk8mk8PBwHThwQOfOndO1a9dUpEgReXl56cEHH1Tz5s1JGuTQqlWrFBISoq1bt+ro0aNKTU295/alS5fWv/71L73++utceAVwTwkJCfr88881f/58nTx58q7bFS5cWC1btlTPnj31+uuv2zFCwDr/+9//9PHHH2vVqlVKSkq663ZVqlTRwIED9frrr3PxOpu2b9+uyZMnKyQkRCkpKVluU7duXY0ePVovv/yyXF2Z7EySzp8/rx07dmj79u3asWOHdu3apdjY2IzXH3jgAYWHh+d4P5cvX9bkyZO1ZMkSxcfHZ7lN06ZNNXHiRD399NM53h9gL3FxcTp06JCOHj2q6OhoJSYmytPTUxUqVFCzZs1Uo0YNR4eY63LrOHLmzBl9/fXX2rx5s3bt2qUrV67ct02LFi00ZMgQ9evXT4UKcUkbAID7cTGZTCZHBwEA+Z29Lq4URFevXtUPP/yg33//XevXr7/niWHhwoXVpUsXvfHGG/L397djlM6jatWqOn/+vNXtChcurPHjxysoKIinnGzshRde0Pfff2+o45hivXfffVeTJ0/Odvt+/fppyZIltguogAkJCVH//v2tOr54e3vrwoULuRhV/te/f38tXbrUJn1xXLFcenq6xo8frxkzZigtLc3idt7e3lqyZIk6deqUi9E5l9TUVI0aNUqffvqpxW38/f21cuVKlS9fPhcjy7u2bt2qmTNnavv27YqMjLzntrb4/z40NFS9evWyKHkjSS+99JIWLFggd3f3HO03r7HH+WBycrL27t1r2M+JEyd052W9xYsXq3///jnaT36Qm+O9Y8cOrVmzRn/99Zf+97//KT09/a7bPvDAA3rllVc0dOhQeXl5ZWt/eZE9jiNffvmlBg8enK34GjVqpGXLlunhhx/OVvu8ytHXlQ4cOKCmTZua3WjlrMeV3BzvnF4TCQsLKxA3mADIfdwuBQDZZM1JEbJnxIgR+vLLL5WcnGzR9ikpKfrhhx/0ww8/6KWXXtKnn36qUqVK5XKUzq9o0aKqXr26SpcurfT0dF25ckURERGGi00pKSmaPHmyzp49q4ULFzowWufy008/mSV/gfzmiy++0PDhw80uoLq7u6ty5coqX768kpKSdOHCBV26dMlBUcLZkjG5aejQofryyy/N6osVK6ZatWrJw8ND0dHROn36tOG78uLFi3r66ae1Zs0aPfXUU/YMOV9KT0/Xs88+q59++snstUqVKqlKlSqKj4/X6dOnDU9gb9y4UY8//rg2b94sT09Pe4acJ+zcuVNr1qyxy762bNmip556Sjdv3jTUe3p6ysfHR1evXtXZs2cNN0p89dVXiouL06pVq/L9TYP2Oh986623tGnTJu3du9fi8yJnlNvjvXfvXvXo0UOnT5+2uM2ZM2c0btw4zZ49W4sXL3aaG3zseRzJrFKlSipXrpyKFy+uuLg4hYeHKy4uzrDN3r175e/vr7/++kuNGjVySJy2kleuK6WlpWngwIF3nWXDWeSV8QYAe2FeJADIptsnRfxozD3bt2/P8iKHm5ubqlatqqZNm8rPz0+lS5c22+arr75Sx44dzU4WcX+VK1fW4MGDtWzZMp08eVLx8fE6duxYxl2x4eHhio6O1vz581W1alVD20WLFmnx4sUOity53LhxQ8OGDXN0GECOfPfddxo2bJgh+duiRQsFBwcrOjpaYWFh2rFjh/bt26eLFy/qwoUL+vrrr9WzZ08SknbWtWtXR4eQL6xatcos+VuvXj398ssvun79uvbv36/t27fr5MmTunjxoiZPnqwiRYpkbJucnKz+/fvr6tWr9g4935k0aZJZ8rdLly7au3evIiMjtXPnTh0+fFiXL1/W559/bnj67uDBgxo4cKC9Q87zSpQoYbO+rl69queff96Q/H3ggQe0Zs0axcTEaPfu3QoLC1N4eLiGDh1qaLt69Wp9/PHHNovFUex1PrhgwQLt2LGjQCd/pdwf73Pnzt01+Vu6dGk99NBDatGihWrWrGl288KFCxfUpUsXfffdd7kSW15iy+OIm5ub2rdvrylTpmjTpk26fv26IiMjtX//fv399986cOCAbty4oR07dqhPnz6GtjExMXr++efvuQRDfpBXrit9/PHH2rlzp0NjsIe8Mt4AYC88AQwAuaBEiRIkHm3M09NTvXv3VpcuXfTYY4+pZMmSGa+lpaVp8+bNmjRpkjZv3pxRv2PHDvXv31+rVq1yRMj50q+//qoGDRrc94kMLy8vDR48WD179lRAQIB2796d8dqECRPUr18/1t/LoTFjxmRMl1u8ePG7rqmH7JkxY4YaNmxo8faVK1fOxWic07lz5zRkyBDDE5BTp07VuHHj7nqM8fb2Vu/evdW7d28SZBYYO3as2QVRS5w4cULDhw831Dnj1H65IfNU8s2aNVNoaKiKFy9utm358uU1adIktWnTRk8++aRSU1Ml3Vovdd68eRo3bpxdYs6PTp06pQ8++MBQ9+qrr2rOnDlm25YsWVLDhw+Xv7+/2rVrlzEV8erVqxUaGqp27drZI+Q8p2TJkmratKmaN2+uFi1aqHnz5goLC1P79u1t0v9HH31kuIDu4+OjLVu2mH1fVq1aVfPmzVP16tU1YcKEjPr33ntPAwYMcKppc+9kj/PBQoUKyc3NLd8nwGwhN8b7kUceUZ8+fdS+fXvVq1fP8Nrly5e1YMECTZ06VQkJCZJuzVrw0ksv6aGHHlLjxo1tGouj5OZxpFOnTrp48aLKli17z+1cXFzUvHlzLVu2TAEBAYbfK8ePH9eiRYuc9qZZe11XOnXqlIKCgjLKBfXcM7fG28/PTzNnzrSqTcWKFW0eB4CCiQQwAORQbl9cKehq1Kihd955R71795aHh0eW27i5ualdu3YKDQ3V8OHD9cUXX2S8FhwcrPXr1+vxxx+3V8j5mp+fn1Xbe3l5afny5apfv35GkicqKkpbt27VY489lhshFgihoaEZT5i5uroqKChIY8eOdXBUzqVp06YFNilgLyNGjDCsoxUUFKTx48db3N5ZkwK2VK9ePbOL0pZYv369ody4cWOrj/8F0enTp3Xw4EFD3dy5c7NM/t7p8ccf18CBAw2/T3766ScSwPfw0UcfGaYNbtKkiWbNmnXPNvXr19d///tf9erVK6Pu3//+t/75559cizMv6tatm5544gnVrVvX7Ga8sLAwm+zj8uXLZusyL1iw4J43S40bN07r1q3Tpk2bJEnXr1/XjBkzNHXqVJvE5Ej2OB90cXFR7dq1Dfto0qSJOnXqpI0bN9psP/lBbo63q6urevfurX//+9+qX7/+XbcrX768xo8fr65du6p9+/aKiYmRdGtZnDfeeCPf/5vY4ziSeSYpS/Tr10+///674UnrVatWOUUC2FHXlUwmkwYPHpxxI0O3bt1048aNfP8Zvh97jreXl5cCAgJs3i8AWIIEMABkkz1Oigq6yZMnq2PHjhZPA+rq6qrPP/9c//vf/7Rr166M+oULF5IAzkW+vr5q2rSpYcyPHDlCAjibbt68qcGDB2ck1F999VU1b97cwVEB1tmwYYPWrl2bUfbz89M777zjwIhwW3p6upYtW2ao4+lfyxw7dsxQrlq1qsXH5x49ehgSwCdPnrRpbM7mxx9/NJT//e9/q1Ch+1++6Nmzp3x9fXXkyBFJt5YT2bt3b75fI9IatWrVyvV9fPfdd4anpNq2basOHTrcs42Li4uCgoIM2y1atEhTpkzJt2sB2+t8cO3atfLz8yuQa1rfKbfHu06dOtq/f/89E7+Z+fn5afHixXr66acz6jZt2qSTJ0+qdu3aOY7JUexxHMmuPn36GBLAt4/3+ZWjrystWLBAGzZskHTrCdjPP/9cffv2zfX9OoqjxxsA7I25GQEgm2rVqqV69eoxzW0u6tKli9VrQLq5uZk9Kblu3TpbhoUsZL5IcHv6RVhv4sSJGYmBatWqacqUKQ6OCLDeggULDOWgoCCLkjfIfSEhITp37lxGuXDhwurdu7cDI8o/bj/hdVu1atUsblu9enVD+dq1azaJyRkdO3ZMFy5cyCi7ubnpqaeesrh9t27dDOU1a9bYLDbckjlBb+l6y+3bt5ePj09G+cKFC/n6CW17nQ+2bdu2wCd/pdwf7zp16liV/L0tMDDQbDaO33//3VZhIRNnO+905HWlyMhIw7WTqVOnWvXbJj/iOh6AgoajHQDA6WR+8jQ6OjpjSiPkjsTEREOZi1TZs2vXLn3yyScZ5blz56pEiRIOjAiw3tWrVw0Jl/Lly5slZOA4S5cuNZS7du2qcuXKOSia/KV06dKG8s2bNy1um3lbxvzuIiIiDOXatWvfd5rtO2V+2jdzshI5ExcXlzGN820dO3a0qK2Li4vZNJg///yzzWIDHCXz+Wfm4xhsh/NO2xk2bJiuX78uSWrRooVGjhzp4IgAALZGAhgA4HSyWjfy9okNbM9kMmnnzp2GuqZNmzoomvwrJSVFL7/8csaah7169VLXrl0dHBVgvb/++stwcS4gIECFCxd2YES47caNG2ZPQzL9s+UyJxaPHDmi+Ph4i9ru2LHDUG7RooXN4nI20dHRhnKZMmWsal+2bFlD+dChQ0pOTs5xXLjl0KFDSklJySj7+PioUqVKFrd/9NFHDeW9e/faLDbAUTKff3LumXu2b99uKHPemT3fffddxnIthQoV0vz583kqFgCcEEd2AIDTOX/+vFld5ouBsJ1FixYpMjIyo1y3bl0ubGfDtGnTdODAAUm37mSfM2eOgyMCsifzDSGtW7fO+O9t27ZpyJAhatCggTw9PVW8eHH5+PioU6dO+vjjjxUVFWXvcAuUFStWGJ5ErVChglVT6xZ0VatWNXyek5KSLDpWJyUlGWZ3kCyfMrcgcnNzM5Rv3xhlqTuTk5KUmpqqEydO5Dgu3JJ5vc3MU9/eT+bt8/v6nYBkfv7JuWfuuHnzpmbNmmWo69evn4Oiyb+io6P12muvZZRHjx6thg0bOjAiAEBuYSEuAIDT2bx5s6H8wAMPWL2WMCyzdOlSDR8+PKPs6uqqzz77TC4uLg6MKv85fPiwpk6dmlGePn26Klas6MCICoakpCSdPn1a0dHRKly4sMqWLavKlSurWLFijg4tX9u1a5eh7Ofnp+joaA0ePDjLtTjDw8MVHh6udevW6Z133tGoUaNYMziXLFmyxFB+8cUXGWcrTZ8+Xf7+/kpPT5ckTZo0SZUrV77rBehr166pb9++hiRXt27dmBb9HjI/8Xvp0iWr2me1/ZEjR7K1tifMHTt2zFC2dr3IzNufOXNGiYmJKlq0aI5jAxzBZDJpy5Ythro6deo4KBrndf78efXt21fHjx/PqGvXrp2ef/55B0aVP73++uu6fPmypFtr4gYFBTk4ooIhKipKkZGRio+Pl5eXl8qVK2fVDBoAkB2c7QMAnM6iRYsMZZ5uyr7jx48b1rBKSUnR1atXdfDgQf344486fPhwxmvu7u6aP3++OnTo4IhQ86309HQNHDgwY3rKNm3aaPDgwQ6OyvmNGDFCp0+fNltHrFChQmratKk6d+6s4cOHq3z58g6KMP86efKkoVy4cGE1b95cYWFh922bkJCgKVOmaOfOnVq1ahVrYNvQyZMntXXrVkMd0z9br02bNvrss880YsQImUwmpaamqn///vr888/17LPP6qGHHpKHh4euXLmi7du365tvvlFMTExG+44dO+rbb7914DvI+2rWrGkoh4eH6/LlyxYfjzPPQiBJFy9etElsME+wV61a1ar23t7eKlSokFJTUyXd+h0UHR2tKlWq2CxGwJ5CQ0MNv3FcXFzUqVMnB0aUP6Wmpio0NNRQFxcXp3PnzmnLli1au3atYRaTRx55RKtXr+bGYyv99ttv+vrrrzPK8+bNk4eHhwMjcn4HDhxQzZo1szwXqlixovz9/dW/f3+OGwByBQlgAIBT+fXXX7Vp0yZDHRe4s2/u3LmaPXv2Pbe5fZFj2rRpTB2VDXPmzNE///wj6f+T6FzIyH133rxwp9TUVG3fvl3bt2/X9OnTNWbMGAUFBZlNSYq7u3btmqE8bNgwwwWPp556Sl27dlX16tUVHx+vPXv2aPny5Tp37lzGNuvWrdPLL7+sFStW2C1uZ7d06VJDuUmTJvLz83NQNPnbsGHD9NBDD+m1117ToUOHJN1KOmaVeLytZs2aGjt2rAYPHswae/fh4+OjqlWrGo4J33//vUaOHHnftsnJyVq9erVZfVxcnE1jLMgyj2Xx4sWtau/i4iIPDw/FxsbetU8gv0hPT9e4ceMMdZ06dWImn2yIi4tTx44d77udt7e3Ro0apTfffFOFCxe2Q2TOIzY2VkOHDs0o9+3bVwEBAQ6MqGCIiYkx3Ax4pwsXLuj777/X999/r8aNG2vp0qVq0KCBnSME4Mw48wQAOI2YmBjDCY0kde/enfVoc1mvXr00YcIEkr/ZEBYWpnfeeSejPG7cOPn6+jowItzp5s2b+s9//qOAgAAuTlsoPT3dcFFfkvbt2yfp1trWf/31l3755RcNGzZMXbp00XPPPadp06bp6NGj6tu3r6HdypUrtWzZMrvF7sxMJpPZWHJzVM48/vjj2rlzp8aMGXPfG0SqV6+uMWPGqHfv3iR/LfTMM88Yyh988IHZzSVZmTVrVpZP+3IMt53MY5mdqZszP23Gvw/yqxkzZmj79u0ZZVdXV8OyLrAtb29vTZgwQUOGDCH5mw1vv/22zp49K+nWOtWZ11OGY+3Zs0ctW7bUypUrHR0KACfC2ScAwCmkp6erT58+hqdFSpcurTlz5jgwqoJhxYoVatOmjdq2bWs29SvubciQIYqPj5ck1a1bV+PHj3dwRM7NxcVFrVu31tSpU/Xnn3/q3LlzSkhIUGJios6fP6+ffvpJQ4cONbuYHRoaqhdeeEFpaWkOijz/iI+Pl8lkMqt3cXHRjz/+qMcffzzLdsWLF9eSJUvMpj57//33s+wP1lm/fr3OnDmTUXZ3d1fv3r0dGFH+N2/ePNWqVUszZsy477EhIiJCw4cPV40aNcyWqUDWRo0aZVif+vz58+rRo8c9E4XBwcGaNGlSlq/dOW0ocibz0gnu7u5W91GkSBFDmX8f5EebN2/WhAkTDHVvvPGGGjdu7KCInN/Fixf12muvqXr16po5cya/Ea2wefNmzZs3L6M8a9YslStXzoEROb9y5cqpf//+Wr58ufbv36+YmJiMJbX27dunzz77zOwm+ps3b6pPnz5ms9oBQHaRAAYAOIW33npLv/32m6Huiy++ULVq1RwUkXP45JNPZDKZMv4SEhJ09uxZ/fzzzxo4cKDhCY7NmzerefPm2rVrlwMjzj8WLlyokJAQSbeSY/Pnz8/WRVRY5oknntDRo0e1detWjR8/XgEBAapSpYo8PDxUpEgRVa5cWV27dtW8efN04sQJPfroo4b2v/zyi+bOneug6POPu60h9tJLL6lt27b3bOvq6qq5c+canpA8evQoF0BsIPP0z127dlXZsmUdFE3+lpKSop49e2rYsGGKioqSJJUpU0aTJk3Sjh07dPXqVSUnJysyMlJr167VM888kzGtf0xMjAYOHKi33nrLkW8hX6hRo4ZZMnf9+vWqX7++5s6dq7CwMCUlJenatWsKDQ1Vnz591KtXL6WkpEi6dRPgnVhP3HYy3ySVnJxsdR9JSUn37BPI606fPq1nn302Yy1rSWrUqBFP/+aAp6en4bwzLS1NMTEx2r17t+bMmWOYFjc2NlZjxoxRv379lJ6e7sCo84fExEQNGjQoI2EeEBCgl156ycFRObfly5fr/PnzWrx4sV588UU1aNBAXl5eKlSokDw9PeXn56cRI0Zo7969mjdvnuHGqOTkZPXu3dvshisAyA4SwACAfG/OnDlm0xeNHTtWzz//vIMicl4eHh6qWrWqunTpoi+//FL79+9Xo0aNMl6/du2aunfvbtE0jQVZVFSUxowZk1EeNGiQHnvsMQdG5Pxat26tOnXqWLRt1apVFRISolatWhnqp0yZooSEhNwIz2kUKlQoyxsZBg8ebFF7Hx8fs/XfNm7caJPYCqq4uDizNVGZ/jn7hg0bpuDg4IxyixYtdOjQIU2ePFnNmzeXp6enChcurEqVKqlbt25avXq1fvjhB0OCa8aMGVqyZIkDos9f3nnnHbPfchERERoxYoRq1qypokWLysvLS+3bt9fXX3+dcWG7a9euevbZZw3tPD097Ra3s8ucTM/OBerMT/ySoEd+cuXKFXXu3FlXrlzJqPP29tbq1au5mcGGXF1d5eXlpcaNG+vVV1/Vvn37NGPGjIybqiRp2bJlmjlzpgOjzB+CgoJ0/PhxSbfO5+98Ehi548UXX7T45u6hQ4fqm2++MdwEe/78eX3++ee5FR6AAoQEMAAgX/vmm2/0xhtvGOr69++vDz74wEERFSy1a9fWn3/+aXjS+vz58/roo48cGFXeN2LEiIwkecWKFfXhhx86OCJkVrRoUX311VeGKUgvXbqkP/74w4FR5Q+ZL+QXLVrUqrXY/f39DeWdO3faJK6CauXKlRlTzUu3LlJ37tzZgRHlX6GhoVq4cGFGuUKFCvr5559VsWLFe7YLDAw0u4g3ZswYpr29DxcXF3377beaOHGixWs9DhgwQN9//73ZOsAkgG0n8zH+zuOLJUwmEwlg5FuxsbHq3LlzRjJNujXjwLp16+Tj4+PAyJyfi4uLRo8erWnTphnqJ0+erKtXrzooqrxv9+7dhpvlJ02apFq1ajkwImTl2WefVd++fQ11y5Ytc1A0AJwJCWAAQL71888/q1+/foa1f5599ll9+eWXhjuDkbvKlSunyZMnG+p4sunuVq5cqTVr1mSUZ8+ezYXpPKp27doKDAw01JEAvj9vb29D2cfHx+LkjSSzJ7UvXbpkk7gKqszH4xdffNFwYwMsN2fOHEP5jTfeUPny5S1qO2DAAMNnOzo62uzJbJhzcXHRe++9p2PHjmnEiBFZJlgKFy6swMBA/f7771q0aJGKFStmWPNaunU8h21UqFDBUD537pxV7S9evGiYNtfV1ZV1KJEvJCYmKjAw0LDcTbFixfTzzz+breOJ3DN69GjVrFkzoxwfH69Vq1Y5MKK8KzU1VQMHDsw45vr5+RlmoULeMnr0aEN5//79Zje0AYC1SAADAPKlDRs2qFevXoYLSB07dtS3334rNzc3B0ZWMN25xqEkRUZGml18xS13rv341FNP6bnnnnNgNLifDh06GMrHjh1zUCT5h6+vr6FcqlQpq9pn3p6nOrIvLCxMmzdvNtQx/XP2mEwmrV+/3lDXrVs3i9u7uLioS5cuhjrWt7acj4+PPvvsM50+fVqRkZHas2ePNm7cqKNHj+r69ev68ccf9eSTT0q6lQw4evRoRls3Nzc1btzYUaE7nYceeshQjoiIsKp95u0feOABps1FnpeamqrnnntOoaGhGXXu7u5avXq12rRp47jACqBChQqZ3aC5bds2B0WTty1fvlx79+6VdOtmm/nz53MTYB7WoEEDw01WJpPJMNsAAGQHCWAAQL6zfft2BQYGGtYca926tdasWWPxOiuwLU9PT5UpU8ZQd+HCBQdFk7fduT7yr7/+KhcXl/v+tW/f3tDHmTNnzLa5fXIP27pzenNJunz5soMiyT/q1atnKCclJVnVPvN6ksWKFctxTAXV0qVLDbNkNGnSRA0aNHBgRPnX1atXdf36dUOdtdN9Zt7+/PnzOY6rIKpUqZIaNWqktm3b6qGHHpKHh4fh9f/9739KS0vLKPv6+qp48eL2DtNp1a1b11A+fPiwVe2PHDlyz/6AvCY9PV19+/bVTz/9lFHn5uamr7/+OuPGE9hX5imMOe/M2p3nnenp6XrkkUcsOvfcuHGjoZ8BAwYYXu/evbu930qBUbVqVUOZc08AOUUCGACQr+zfv1+dO3dWXFxcRl3jxo3166+/cnEvj7Fmylcgr8r8OU5JSXFQJPlHkyZNDGVrpy7LPOVz2bJlcxxTQWQymfTVV18Z6gYMGOCgaPK/rG5ksPYpmszHkzuTlLCd4OBgQ5k1r22rfv36hs9yeHi4oqKiLG6/detWQ7lRo0Y2iw2wNZPJpCFDhui7777LqHNxcdGXX36pnj17OjAy3InzTjgLzj0B2BoJYABAvnHs2DF17NjRMB1o3bp1tW7dOpUuXdqBkSE2NlYxMTGGuszrgAL5UeYnCixd77Mge+KJJwzTeUZFRVmVHNi9e7ehnHm6UVhm48aNCgsLyyi7u7vrX//6lwMjyt+yuhEhMjLSqj4yP/HL8cT2kpOTDYkaSRo4cKCDonFOJUuWVNu2bQ11f/75p0VtTSaTQkJCDHXWTKUO2Nubb76phQsXGurmzJnDcgoOlnmpIc474Sw49wRga0z8DwDIF86cOaOAgADDk2E+Pj4KCQnhR3Ee8MsvvximGS1fvrwqVarkwIjyrh9//NHqO3n37dunMWPGZJS9vb21fPlywza1a9e2SXww2rJli6GceUpomCtevLg6duxomCoxODhYI0eOvG/btLQ0/fDDD4a6du3a2TrEAmHp0qWGcrdu3XiaOgfc3d1VqVIlw80M69evtyq5+NdffxnKmaewRM7NmjXL8FvR39+fm0hyQWBgoOHzvHDhQr300kv3bbdhwwbDjSne3t5q2bJlrsQI5NTEiRM1e/ZsQ937779v0e8Z5B6TyaSff/7ZUOfn5+egaPK2Hj166OGHH7a63ejRo7V///6M8lsQK3o+AAAgAElEQVRvvaUnnngio3znOrWwnXPnzpnd3MC5J4CcIgEMAMjzoqKi1KFDB507dy6jrnLlygoJCVGVKlUcGBkk6ebNmwoKCjLUde3aVa6uTDSSFX9/f6vbZJ5mtGjRogoICLBVSLiLa9euafXq1Ya6Dh06OCia/GXIkCGGBPDMmTP18ssv33c934ULFxqeqixVqhTr62VDfHy8Vq1aZajjaaWc69Chg+Hmm08++UT9+vWzaCrojRs36u+//zbrD7Zz8OBBTZ06NaPs6uqqjz76yIEROa8XXnhB48ePV3x8vCRp06ZNWr9+vR5//PG7tjGZTJo8ebKhbsCAAfxeRJ700UcfacqUKYa6cePGady4cQ6KCLctXrxYR48eNdQFBgY6KJq8rVq1atlKIHp5eRnK9erV49zTDjLPNlCtWjU9+OCDDooGgLPglzYAIE+LiYlRx44dderUqYy68uXLKyQkRDVr1nRgZM5n7Nix2rlzp1VtYmJiFBgYqOPHj2fUubm56Y033rB1eIDdjRkzxjDlvLu7O2tJWqhr16569NFHM8rh4eEaNGiQ0tPT79pm165deuuttwx1r7zyClP8Z0NwcLDi4uIyyt7e3urUqZMDI3IOffr0MZQPHjyo4cOH33ct35MnT6p3796GugcffFCtWrWyeYzOJCIiQjdv3rRo2z179qhjx46Gz/3IkSPVvHnz3AqvQKtQoYLZU5CDBg2657To06ZN06ZNmzLKpUqVMjvmA3nBF198obFjxxrqRo4cqffff99BETmfjRs36sMPP1RCQoJV7VauXKlhw4YZ6nr16qUHHnjAluEBdnfkyBHNnDnTUNe9e3cHRQPAmfAEMAAgz4qNjVWnTp106NChjDpPT0+tW7dOvr6+DozMOf3xxx/66KOP1KJFCz3//PN6/PHHVb9+fRUuXNiwnclk0rFjx7Ry5UrNmTNHV65cMbz+5ptvMg0X8pQPPvhAHTt2VNOmTS3aPjU1VW+//bbZXdivvPIKU5tb4eOPP1br1q2VmpoqSfr2228VFRWlDz/80JCUuXHjhhYvXqyJEycqNjY2o7527dqaMGGC3eN2BkuWLDGU+/TpY9FTqri3J598Uu3bt9eGDRsy6hYsWKAjR47o3Xfflb+/v2Gco6OjtXTpUr333nu6fv26oa/3339fbm5udos9P1q7dq3effdd9enTR88++6xatGhhWF9cupX4XbJkiebOnZtxrJGkxo0bF+hkzdatW7NMnu/bt89QTkxMNFuT97bKlSurXr16d93H2LFjtXTp0oz1CsPCwtS6dWvNnj1bgYGBcnFxkXRrSsspU6boiy++MLR/5513VKZMGaveF5DbvvnmGw0fPtxQN2DAAM2ZM8dBETlObh5Hrl69qrffflvTpk3Ts88+q2eeeUbNmzfPci3f2NhYbdiwQV988YV+/fVXw2teXl5mSTPAkfbu3asNGzZo6NCh95356M42Tz/9tOE8yMPDQ2+//XZuhQmgAOEqAADkgD0urhRkgYGBZk+kjho1StHR0Xcdz7tp2rSp2VRGyNqOHTu0Y8cOSbeeeKxSpYo8PT3l7u6u2NhYnT171nBycqd+/fpp+vTp9gwXuK/ff/9d48aNU+vWrfXcc8+pQ4cOqlu3rllC7Pr16/r111/14Ycfau/evYbXatWqpUmTJtkz7HyvefPm+vTTTw1PaoSGhqpFixby9vZWtWrVlJCQoJMnTyo5OdnQ1svLS6tWrVKpUqXsHXa+d+bMGYWGhhrqmP7Zdr755hu1bt3asI7pli1bFBAQoBIlSsjHx0ceHh6Kjo7W6dOnZTKZzPoYPXq0evbsac+w863o6GjNnj1bs2fPVqFChVSjRg2VKVNG8fHxioyMNMzScJufn59+++03FS9e3AER5w0vvvii2TqCWbl48aI6duyY5Wv9+vUzu5nkTmXKlNH333+vJ598UomJiZJuHX+6d+8uT09P+fj46Nq1a4qIiDB7Sv7pp5/W6NGjLX9DeZg9zgejoqIMN8TeKfP/A4cPH77rftq0aWN2E0V+k5vjHRISon79+hlmK6lbt66ef/55szXc78fLy8viGw/zKnscR65du6ZFixZp0aJFkm7N9FWuXDmVKlVKycnJiomJUURERJbfpZ6engoJCXGKNVK5rmRfuTne165d06hRozR16lTDzQ3lypUzbGcymXTw4EEtWLBA8+fPV1JSkuH1adOmsdwZANswAQCy7YEHHjBJytFfv379HP028qycju2dfxs2bHD028nzGjZsmO3xLVWqlGnu3Lmm9PR0R78Np7RhwwbDeD/wwAOODilf8ff3N/vMFilSxFSrVi1TkyZNTM2bNzfVrFnT5OrqmuXnu2LFiqbjx487+m3kWwsXLjQVLVrU4uPJgw8+aDp69Kijw8633nvvPcN4Nm3a1NEhOZ2IiAhTu3btrP6uLFy4sOmDDz7gu9JCn376qdVj3LdvX9ONGzccHbrD2fMc5a+//jKVKVPG4n579+5tSkxMzN0BsCN7jPXixYttcj4UFhZmlzHJTbk53kFBQTYZZ0kmf39/u45LbsjNsV6zZk2O+u3YsaPp9OnT9h2QXJTXritlPndavHixzfrOC3JzvDOft9/+8/b2NtWvX9/0yCOPmB5++GGTl5fXXfsePXq0fQcEgFNjDWAAACDp1vSs06dPV0BAgEVP3bm4uMjPz08fffSRTp48qWHDhmVM9wfkdUlJSTp16pR2796tnTt36vTp01muT/vUU09p3759evDBBx0QpXN4+eWXdfDgQfXp00fu7u533a5GjRqaNWuWDhw4oIceesiOETqXr776ylDm6V/bq1atmv766y+tWLFC7dq1k6vrvU+rS5curWHDhunAgQN6++23+a60ULt27dSvXz9VrFjxntu5u7vrmWee0datW/XVV1+pZMmSdooQkvT444/r8OHDGjZs2D2nu2zcuLGCg4P19ddfq0iRInaMEEBe88QTT2j16tUaPHiwHnroIYu+F0uVKqXevXvrr7/+0h9//CEfHx87RArYxsWLF3Xo0CH9888/OnjwYJYzmJQqVUrLly/XjBkzHBAhAGfFFNAAAECS5OvrK19fX40dO1bp6ek6ceKETp48qYiICN24cUMpKSkqWbKkSpcurRo1aqhJkyZMz4p8YcKECfL19dXmzZt19OhRs6koMytRooQ6d+6skSNHqm3btnaK0rnVqlVLy5Yt03//+19t27ZNx44d040bN1S8eHFVqFBBjRs3Zm13G9iyZYtOnjyZUXZ3d1fv3r0dGJHzcnV1Va9evdSrVy/FxsZq165dOn36tK5du6bExESVKlVKZcqUUcOGDVWvXr37Jolh7uGHH86YOvT06dM6ePCgIiIidP36dbm4uMjLy0t16tTRI488UqCne85KeHi4Xffn7e2tuXPnaubMmdq2bZuOHDmia9euZSwl0rJlS9WuXduuMQHImdw8jhQrVkzPPPOMnnnmGUm3ps09fPiwwsLCdOnSJcXHx8vd3V2lS5dW2bJl1aBBA9WpU4cbqJDnNWjQQNOnT9eGDRu0Y8cOxcTE3LdN3bp19fLLL2vQoEEsWwbA5lxMpiwWUgAAAACcUEJCgg4fPqzw8HBFRUUpLi5O6enp8vT0lJeXl+rVq6cGDRrIzc3N0aECAAAAAPKpM2fO6MSJE4qIiNDVq1d18+ZNFS1aVF5eXqpUqZJatmypsmXLOjpMAE6MBDAAAAAAAAAAAAAAOAnmoQIAAAAAAAAAAAAAJ0ECGAAAAAAAAAAAAACcBAlgAAAAAAAAAAAAAHASJIABAAAAAAAAAAAAwEmQAAYAAAAAAAAAAAAAJ0ECGAAAAAAAAAAAAACcBAlgAAAAAAAAAAAAAHASJIABAAAAAAAAAAAAwEmQAAYAAAAAAAAAAAAAJ0ECGAAAAAAAAAAAAACcBAlgAAAAAAAAAAAAAHASJIABAAAAAAAAAAAAwEmQAAYAAAAAAAAAAAAAJ0ECGAAAAAAAAAAAAACcBAlgAAAAAAAAAAAAAHASJIABAAAAAAAAAAAAwEmQAAYAAAAAAAAAAAAAJ0ECGAAAAAAAAAAAAACcBAlgAAAAAAAAAAAAAHASJIABAAAAAAAAAAAAwEmQAAYAAAAAAAAAAAAAJ0ECGAAAAAAAAAAAAACcBAlgAAAAAAAAAAAAAHASJIABAACAPCQkJEQuLi6Gv0KFCjk6rHyvatWqZuO6fPlyR4flVNq0aWM2xlOmTHF0WAAAAAAAFDhcSQIAAAAAFHiJiYk6fvy4wsPDdeHCBcXHxyspKUklS5aUl5eXvLy8VLVqVdWrV09ubm6ODhcAAAAAgLsiAQwAAIA878yZM1q2bJlVbVxcXFS8eHF5enqqdOnSql69uvz8/FS4cOFcihJAfrN7926tXLlS69ev1549e5SSknLfNsWKFVOTJk3UsmVL9ejRQ61atbJDpAAAAAAAWI4EMAAAAPK8sLAwTZw4Mcf9FClSRI0aNVLXrl01aNAgVaxY0QbRAchP0tLS9O233+qjjz7S/v37rW6fkJCgLVu2aMuWLZo5c6Zq166tvn37atiwYSpfvnwuRAwAWUtPT9d7771nVv/yyy+revXqDogIAAAAeQUJYAAAABQYSUlJ2r59u7Zv36733ntPL7zwgj7++GOVLVvW0aEBsIMtW7ZoxIgR2Ur83s3JkycVFBSkGTNmaOzYsRo1apSKFStms/4B4G7S09M1efJks/qAgAASwAAAAAWcq6MDAAAAABwhJSVFy5Yt08MPP6w//vjD0eEAyEW3kyT+/v42Tf7eKTY2VhMnTtSDDz6opKSkXNkHAAAAAACW4AlgAAAAFGgXLlxQt27d9Pvvv6t9+/aODgeAjaWkpOhf//qXgoOD77ndI488oieeeEItW7ZUrVq1VLFiRRUvXlypqamKjY3VmTNndPToUW3atEnr1q1TRERElv1ERkYqJSVFRYoUyY23AwAAAADAfZEABgAAQL4VFBSkd999966v37hxQ5cvX9aOHTv0yy+/6Pvvv1dqaqrZdsnJyXrmmWe0a9cu1a5dOxcjvr+AgACZTCaHxuCMzp075+gQ4ABpaWnq2bOn1q5dm+XrhQoV0sCBA/XWW2+pVq1ad92maNGiKl++vJo1a6Y+ffrIZDJp06ZNmj17tn744Qf+nwUAAAAA5ClMAQ0AAACnVapUKdWqVUv/+te/tHz5ch0/flyPPPJIlttev35d48aNs3OEAHLTm2++edfkb4MGDbRr1y7Nmzfvrsnfu3FxcZG/v79Wr16tvXv36rHHHrNFuAAAAAAA2AQJYAAAABQYPj4+2rhxo9q2bZvl68HBwdqzZ4+dowKQG4KDg/Xpp59m+VqnTp20bds2NWzYMMf78fPz06ZNmzRnzhymfQYAAAAA5AkkgAEAAFCguLu7a+XKlSpRooTZayaTSStWrHBAVABsKSYmRsOHD8/ytTZt2mjNmjVZHgNy4tVXX9WWLVvk7e1t034BAAAAALAWawADAACgwKlQoYJeeeUVzZgxw+y1P//8U9OmTXNAVHAmx44d08GDBxUVFaW4uDh5eHiocuXKatiwoerUqePo8Jze+++/r0uXLpnVly9fXitWrFDRokVzZb/NmjXTP//8w5PA+Vx6eroOHTqkw4cP6+LFi0pISFCxYsVUvXp1NWnSRNWrV3d0iAAAAABwTySAAQAAUCB17949ywTwnj17lJiYaJME0d9//61ff/1V//zzj44fP66YmBglJibK09NT9erV09dff62qVavmeD/ZFRkZqVWrVikkJEQHDx7UpUuXlJycrHLlyql8+fJq1qyZOnfurM6dO6t48eK5FsfNmzf1559/atOmTdq9e7fCw8N15coVJSQkqEiRIipZsqQqVqyoevXqyc/PTx06dFDTpk3l6pq3JjS6dOmS5syZo+XLl+vMmTN33a5mzZoaNGiQRo4cqZIlS9ps/+np6dq9e7c2b96sw4cP68iRIzp79qxu3LihuLg4FStWTGXKlFHZsmVVr149tW3bVv7+/nrwwQdtFkNecPHiRc2dOzfL16ZPn65KlSrl6v5r1KiRo/YxMTFau3atNm7cqAMHDujMmTOKjY1VWlqaSpQooSpVqqhu3bp69NFH1b17d/n4+NgmcAskJiZq7dq1+uWXX7Rv3z5FREQoNjZWHh4eqlixopo3b67u3bure/fuKly4sEV9RkVFacWKFQoNDdX+/ft1+fJlJSUlqXz58qpSpYoCAgLUs2dPNW7cOJffnRQeHq5Zs2bp+++/z/IGgtsaNGigoUOHavDgwXJ3d8+VWA4cOKCff/5Z27Zt0/Hjx3XhwgXFx8ercOHC8vT0VI0aNdSoUSO1b99eXbt2VbFixXIljqxcvXpVwcHBWrdunfbv36+oqCjdvHlTZcqUUfny5eXn56fOnTura9eu8vLyyrU44uLi9Ouvv2rjxo3at2+fwsLCdO3aNSUlJcnDw0Pe3t6qU6eOWrdurcDAQPn5+eVaLJnl5vfrnDlzdOPGDUm3jvtZ+eqrrxQaGnrfvlxdXTV+/Hir9g8AAIB8wgQAAADkcRs2bDBJMvsLCgrKdp/JyckmFxeXLPs9e/bsXdtNmDDBbPsOHToYtvnll19Mfn5+WfZ959+BAwfM+v/zzz/NtnNzc7P4fVnSPjo62jRkyBCTu7v7fWOUZKpUqZLpyy+/NKWlpVkchyVOnTplGjZsmKl48eIWxXHnX9myZU2vvvpqlmOYlSpVqpj1sWzZMotjvV/7xYsXm0qXLm3Ve/D29jb98MMPVo/bnRISEkwrV6409ejRw+r93/4LCAgwbdiwIUdxmEwm06OPPmrW93/+858c92utadOmZfk+69ata0pPT7d7PJY6ffq0acCAARb/f3nnv9/WrVtztO/7/dulp6ebPvvsM5O3t7dFMfn4+Jh+++23e+7z0qVLpqFDh5oKFSpkUZ8vvPCCKSoqKlvvLyUlJcs+N2/enPH+pk+fbipSpIhVY1+rVi3Tli1bshXT3axdu9bUrFkzq+IoUaKEafTo0abLly9ne78nTpzIsu87vw/j4+NN//73v00lSpSwKK7SpUubPvzwQ1NSUpIthiZDWFiYaciQISYPDw+rxumRRx4x/fnnn9neb175fs3q+yi7f9b8vgAAAED+krdumQcAAADs5PZTVFm5cuVKtvpMSUnRoEGD1KVLF+3fvz8n4eWarVu3ql69epo/f76Sk5MtahMVFaVBgwapT58+Sk1NzXEMqampmjhxonx9ffXf//5X8fHxVvcRHR2tTz/91C5PBd7P+PHjNWDAAF2/ft2qdhcvXlT37t31n//8J1v7PX36tLy9vdWrVy8FBwdbvf/bQkJC1L59e/Xt21eJiYnZ6iMvWbRoUZb1b775plxcXOwcjWXmzJmjhx9+WIsXL7b4/8vbQkJC1KZNG73yyiu58u9348YNPfnkkxo5cqQuXrxoUZuwsDA99dRTmjVrVpavb9y4UX5+fvriiy8sPqZ89913atasmU6dOmVx7JZIS0vTSy+9pLfffltJSUlWtT116pTatWunJUuW5DiOmJgYPf300woMDNSuXbusahsXF6eZM2fK19dXwcHBOY4lK0eOHFHDhg31wQcfKC4uzqI2169f19ixY9WpUyeL29xLenq6pk2bJl9fX82fP183b960qv0///yjjh07asCAAVa3tURe+H4FAAAAbiMBDAAAgALrbtM8Z+fCcFpamnr06KGFCxfmNKxc88cff6hDhw4WJ3Ey+/bbb9W3b98cxXDlyhX5+/trypQpVie6smIymXLcR0588sknOV4zetKkSfrggw+sbpeQkKDY2Ngc7ftOy5cvl7+/v2JiYmzWp70dOXJEJ06cMKt3d3fXc88954CI7i09PV2DBg3S66+/roSEhGz3YzKZ9MUXX6hdu3a6du2azeKLi4tThw4d9Of/tXfvUVGV+//A3zOA6IAKooCAYF7yLhoqaQhKZnjLS7qko3nXc7DOV9MkQzNLzLS0Yy0s9Zz0eJeOqWmlaQreFTEREZVUQPEG4gW5X/bvD5f9xP1smNkzMAO8X2uxVn72PJ/5MLP3zIrPfp5n3z5VNc2cORMbNmwoFd+7dy+CgoJw+/Ztg3OmpaUhMDBQ9U06IqIaDVFUVIQJEyZg8+bNqnNcu3YNXbt2xU8//aQ6B/Dk83XEiBEm38f+7Nmz6NGjB/78809V4w8ePIigoCAUFxerruHx48cYMGAAwsLCjL7RYe3atejdu7fqm2ZELOH7lYiIiIjoWdwDmIiIiIhqrPv37wvjDRo0MDjXhx9+iF27dpWKabVadO3aFe3atYOLiwsKCgpw48YNJCYmVvoM4XPnzmHYsGGyGW6tW7eGr68vGjVqBFtbW9y5cwdHjx5FYmKiMM+WLVswdOhQVc20p83fCxcuKD7G1tYWfn5+8PT0hLOzM6ytrZGZmYm0tDScPn0aN2/eNPh5K0pcXByWL19eKta0aVOMGTMGgwYNgqenJ+rVq4dbt27h/Pnz2Lx5M7Zt2yZsfIeFhaFLly7o06eP0XVZW1ujVatWaN++PRo0aAAHBwdYW1vj4cOHuHHjBmJjYxX3KD516hQmTpyI7du3G12HOezZs0cY79mzp+KMf3P6v//7vzJvGnFxcUFAQAA8PDxga2uLtLQ0xMbGIiEhQfj4kydPIigoCIcOHTLJ3rSjR4+WzUZ1dXVFQEAA3NzcYGdnhzt37uDIkSOKnxkhISEIDAyEm5sbEhMTMXz4cFkD76WXXkLnzp3h4uKCwsJCpKSkYN++fcLP6NTUVISGhirO9DbEL7/8IruG27Vrh7Fjx6Jv377w8PCATqfDjRs3cObMGWzatAm7du2S3XgiSRLGjx8Pb29vtG3b1qAaMjIyEBgYiOTkZMXH+Pj4oGPHjnBzc8Pjx49x/fp1HDhwQNjslyQJYWFh0Ol0mDZtmkG1iNy8eRNvvPGG7Lm8vLzg5+cHFxcX6HQ6ZGRkICYmBmfOnBHemHP06FEsXboUoaGhBteQk5ODfv364ciRI4qP8fLyQvfu3eHi4oL69esjMzMTycnJOHjwoHCViZMnT2Lw4ME4cOCA0fvJW8L3KxERERGRjBmXnyYiIiIi0ktF7AF8/fp1xT3xMjIyFMeJ9gB2dXWVtFrtX/+2tbWVZs+eLd25c0cxT3x8vHC/xorYA1ir1UqtW7f+698ajUYaPXq0lJSUpJjn5MmTUufOnYWvj6urq1RQUKB3TZL0ZM9lPz8/xde8TZs2UmRkpJSdnV1mnpSUFGnp0qV/7ZGp72tTEXsAP7936fTp06WcnJwy85w9e1Zq37694n6i5Y1/Vnx8/F9jPT09pWnTpknR0dFSXl5euWPj4uKkSZMmKe6DvXLlSr3reMoS9gAeOXKk8Pcxx17E5fnpp58Ur4fmzZtLu3btkoqKioRj//jjD+nVV19VHB8aGmpQLaL3rlWrVqX+3bFjR+m3335T3Ec5KipKatGihbCeSZMmSfn5+VLbtm1LfS5NnDhRSk1NFebLz8+XFi5cKFlZWcnyaTQaKTY2Vu/fT2kP4GevYRsbG2nhwoWKr/lTBw4ckDw9PYX5XnnlFYP3mR42bJji+xgcHKz4OZ2Xlydt2LBBcnZ2Fo6tVauWdPbsWb3rUNoD+Nn3DIA0YMAA6cyZM4p5EhMTpcDAQGGuOnXqlPm9qCQ4OFjx/ZswYYJ04cIFxbF5eXnSypUrFfevXrhwod51WOL3a3n7WxMRERFRzcUGMBERERFZvIpoAK9bt06Y09HRscw/4IsawM//8TY+Pl51XRXRAH72R6fTSbt27dIrV1ZWltS9e3dhnv/9738G/V7z5s1TrOnTTz+VCgsLDconSZIUHR0tBQUF6fXYimgAq20iZGZmypoqT3/CwsL0zhMfHy/5+vpKP/74o8FNp6cOHTokOTo6yupo0qRJuY2w51lCA/jFF18Uvq67d++u1DrKk56ertiQGjJkiPT48eNyc5SUlEgLFiwQ5tBqtQY1gETv3bM/Y8aM0et8SEtLk5o0aSJs/M2YMaPUv/V9T9asWSOsaerUqXr/fkpNsmebdhs2bNA7X3JystS4cWNhrlWrVumdR+l7SKvVSuvWrdMrx927dyVfX19hng4dOuh1Q4gkKTeAn/0e0vd3KywslIYOHSrM8+WXX+qV46m1a9cK87i7u0tHjx7VO8+tW7eETVcbGxspLi5OrxyW+P3KBjARERERKeEewERERERUI61YsUIYDwwMhEajUZXT3t4eUVFRaN++vTGlVRitVoudO3di4MCBej3e3t4eGzZsEO6V/N///lfv57127ZrinpSrV6/GRx99BGtrw3en8ff3x6+//mrwOFMLDg5GWFiY3o93dHTEL7/8Ajs7O9mxiIgIvfeCbdu2LU6cOIGhQ4eqPmd79uyJPXv2wMbGplT8+vXr2Llzp6qc5lJSUoKrV68Kj7Vp06aSqynbkiVLhHuF+vv7Y8uWLcJz43kajQZz587FzJkzZcdKSkowa9Ysk9Q6bNgwrF27FlZWVuU+1s3NDcuWLZPFc3NzS8UjIyMxYMAAvZ5/3LhxCAoKksW3bt2KwsJCvXKU54MPPsCoUaP0fryXlxd27twpXDr4iy++0CtHUVERZs+eLTwWERGh936wjRo1ws8//4xWrVrJjsXHx2Pt2rV65SnP6tWrMXnyZL0ea21tje+//x7Ozs6yY4Z8d2RlZeH999+XxZ2dnXHkyBH06NFD71yurq44cOAAvLy8SsULCwuxZMkSvfMoMdf3KxERERGREjaAiYiIiKjGWb9+PU6cOCE81rdvX9V5Fy5cKPwjvKWYOXOmwXvMNmvWTNgYKWsvxueFh4cLGzUhISGYNGmSQfVYGp1Ohy+//NLgcV5eXvjggw9k8YcPHyIyMlKvHMbuW/lUt7rxOW0AABqTSURBVG7d8M4778jiGzZsMEn+ynLnzh0UFRUJj3l4eFRyNcry8vKE+9fWqVMH69evh62trUH5Pv/8c7Rr104WP3HiBGJjY1XXCTxptK1atcqgGwzefPPNMl/vyZMn690ke2rGjBmy2L179xT3QjaEm5sb5s6da/C4rl27Yty4cbJ4UlISoqKiyh2/fft24Z7m/fv3xz/+8Q+DanFyclLcEzkiIsKgXCIjRozA+PHjDRrj4OCAf/7zn7J4fHy8cO9ikW+++QYZGRmy+A8//ICmTZsaVM/TmtatWyeLR0ZG4tatWwbne5a5vl+JiIiIiJSwAUxERERENcrPP/+Mv//978Jjzs7OBs0Ce5a7u7uwiWYpdDodPvzwQ1Vjg4ODZbH79+8jOTm53LEPHz7E5s2bZfEmTZroPVPOkk2YMAHu7u6qxs6cORP29vayuFIjpyKJZvYp3SRhqe7evSuMOzg4oFatWpVcjbItW7bg3r17svisWbPg6elpcD5ra2vhrFvA+Obfu+++CycnJ4PGaDQavPHGG8JjVlZWmDNnjsF19OrVC/Xq1ZPFz549a3Cu582YMUOvGdci8+bNEzbH9bmGRe+NlZUV/vWvf6mqpUePHnjrrbdk8fj4eBw6dEhVTuDJ+/npp5+qGiv67gD0e99KSkqwcuVKWXzEiBHw9/dXVQ/wZJb98+MLCwuNuuHFXN+vRERERERlYQOYiIiIiGqEa9euISQkBIMGDUJubq7wMXPmzFHdCBgzZoxeS6SaS3BwMBwdHVWN7dq1qzCuz+y7n376Sfh6T506VfVrbUn+9re/qR6r0+kwePBgWfzkyZPIz883piyDtW3bVrZc661bt3D9+vVKrcMYSktnW9p5tmvXLllMq9ViypQpqnP27dsXzZs3l8V3796tOicA1TP0vb29hfHXXntNtgSvPmxsbISznI2dAazRaBSblPrw8vISLkMcHR1d5rhHjx7h8OHDsnifPn3QsmVL1fWEhIQI46JzTl+9e/dG69atVY1t0aKF8HtHn/ftyJEjSE1NlcWnT5+uqpZnjR07VhYTvR/6Mtf3KxERERFRWQzfaIuIiIiIyEIcOnQI4eHhisezsrKQnp6OmJgYJCQkQJIkxccOHDjQqBm8+u5naS6BgYGqx9avXx9ubm6y5UozMzPLHbtnzx5ZzNraGhMmTFBdj6Vo1KgRunfvblSOIUOGYOPGjaViBQUFiIuLQ7du3YzKbajGjRvLZtEmJCSgSZMmlVqHWnl5ecK4Jc3+BZ40+J/Xq1cv1TPJnxo1apRspmZ6ejquXLkibA6Xp1WrVmjcuLGqWl588UVhPCAgQFU+AGjZsiWOHz9eKiaaSW0IHx8fo1/3IUOG4OjRo6ViqampuHv3rnAPXODJOVBSUiKLq12B4ik/Pz80bdpUNnvUmNn8xnx3AE/23z527FipmNrvDnd3d6M/c4Ene58/7/lzyxDm+n4lIiIiIioLG8BEREREVGUdPHgQBw8eNDpPjx49sHXrVtUzeLVaLTp16mR0HRVJaZaRvhwdHWV/oH748GG5406dOiWLdezYUbExUpUozXI0RY6YmBijG8DJycm4cOEC7t+/j0ePHiErKwsFBQWKj79//75eMUuldP0qNYbN4fr160hLS5PFjVnS9iml5uqJEydUNYB9fHxU1yJarhkAunTpojpn/fr1ZbFHjx6pzgfAJJ/bZV3DSjcGKTVkjT0PNBoNevbsKWsAx8bGoqioCNbWhv8JyBTfHc/T57tD1JB9+eWXDdqPWknz5s2h1WpLNeEzMjKQmpqqahl2c32/EhERERGVhQ1gIiIiIqqxrKysEBoaik8++QQ2Njaq8zRp0sTilpl9nrENV1FDJysrq8wx+fn5uHLliixe2TNbK0qHDh2MztG8eXPodDrZ8sUpKSkG53rw4AG2bduGLVu2ICYmxiQNhAcPHhido7LUqVNHGLekRsr58+eFcVM0IpVyKD1neYz5zBDtbQ08mTVvypzlfQaVxxTXcMeOHYXxsq5h0Xvi6Oioanns53Xq1Anr168vFcvNzcWVK1fQqlUrg/OZ47sDAM6dOyeLqV2K+nlarRYODg6yWbbp6emqGsDmeo2IiIiIiMrCPYCJiIiIqMaxs7PDpEmTEBsbi88++8yo5i8AODg4mKiyiqHRaISz5wyh1cr/16G4uLjMMTdu3BAuuy3ay7MqcnFxMTqHVqtFw4YNZXFDZt7m5OQgLCwMrq6umDRpEvbv32+ypqclNU/LozTrNCcnB4WFhZVcjZjSsq5qGnPPa9CggbDBqnYpWWM+10SfFxWRs7zPoPKY4hpu1KiRcFZqWdew6D1RWjbbUEpNUnOcB4C69+3x48fCehcuXAiNRmOSH1F+NSsemOv7lYiIiIioPJwBTERERETVlk6nQ/369eHg4ABPT0906dIFXbt2Re/evRWbRWoY+8ffiqbVak2ybKahMjIyhHFLb5jry1TnkCiPvjNvExISMGDAAFUzhvVRVFRUIXkrQll7uT58+FDYaK9sSu+rqT5DHBwckJ6eXiqmdhlvtUviV3ZOY5jiGtZqtbCzs8Pjx49Lxcu6hkXHTHkOiKg9D9QsG22s27dvV/pzAupeI3N9vxIRERERlYcNYCIiIiKqsj7++GPMnz/f3GUYPYO4usrNzRXGq0sDWKfTmSSPaPnw55tJIgkJCejdu7es4VdT2dvbw8HBQdhcu3jxIvz8/MxQVWlKDaaKvJmgKu3jXNlMeQ0/f82WdQ2L3pOKPAeUntNSPb8kfmXJz883y/MSEREREVUELgFNRERERESVqrrMljJVkyI7O1sWU9pD9amioiIEBwcrNn/d3d3x9ttv47vvvsNvv/2GCxcuID09HY8ePUJBQQEkSZL9vPLKKyb5fcypffv2wvjp06cruRLDmOqaqC7XVmUx5zX8PJ4D/x+XPyYiIiIiMh5nABMRERERUYWoU6eOMK7v8saW7tGjRxWWp7xZ0t9++y3Onz8vi7u4uGD58uUYPny4wcvtFhQUGPR4S+Tj44MjR47I4jExMWaoRk7pfX306JFJZqOK9mx2dHQ0Om91ZYpruKSkRNgALusaFh0z1eeJ0r7dVek8qF27tjA+evRok+yXraRTp04VlpuIiIiIqLKxAUxERERERBVCac/VqrQUaVnu3r1rdA5JkoR7JZfXAF61apUs5uzsjNOnT8PDw0NVLZmZmarGWZKAgAAsX75cFj9y5AgkSTL77EilJtyDBw/g6upqdH42gA1jims4PT0dkiTJ4mVdw6L3xFQ3xlSHBrBSrf7+/pg8eXIlV0NEREREVDVxCWgiIiIiIqoQHh4ewobbhQsXzFCN6Z07d87oHH/++adwGVpPT0/FMcnJycLZv0uXLlXd/AWAe/fuqR5rKV577TXUqlVLFk9NTcX+/fvNUFFpDRo0EMaTkpKMzv3gwQNhQ7MqNf4qmymuYaUcZV3DovPAFOcAAFy6dEkYr0rngYuLi/A6vnr1qhmqISIiIiKqmtgAJiIiIiKiCmFra4uWLVvK4qdOnTJDNaYXFxdXYTm6du2qOEa0n61Op8Obb76puo6rV69Wi6W57e3tMWDAAOGx1atXV3I1cu3atRPGz549a3RupRxK+yKTaV53Ndew6DzIzMzE9evXja5H9DvVqVMHzZs3Nzp3ZdFoNGjTpo0sXl2+O4iIiIiIKgMbwEREREREVGF8fX1lsbi4OKSnp5uhGtNKT0/H8ePHjcqxY8cOWczGxgadO3dWHHPnzh1ZrHnz5op7Luvj2LFjqsdaGqUlYnfu3Ilr165VcjWleXp6ws3NTRY/dOiQ0bmVcnTv3t3o3NXVmTNnkJaWZlQO0TXcpEmTMpf0VnpPTHEeHD58WBbz8fGBjY2N0bkrk+i74/DhwybbK7m60GrFf9YTLUtORERERDULG8BERERERFRhgoKCZLGioiJ8//33ZqjG9DZt2qR6bE5ODnbu3CmL+/r6wtbWVnGcaKlmY5d3Xb9+vVHjLUlQUBC8vb1l8YKCAkycONHsjZGXX35ZFjt48CBu375tVN6NGzfKYo0aNapSMz8rW0lJCbZs2aJ6fEpKivDmiYCAgDLH+fr6Cht3xnyeAE9u5BAtkyw65yxd//79ZbHCwkJs3brVDNVYLq1WK7z5Jzc31wzVEBEREZElYQOYiIiIiIgqzKBBg2BnZyeLr1ixQrj3bVWzZs0a1TMIly1bhsePH8vi48ePL3Oc6PU0Zv/exMRE7Nu3T/V4S6PRaBAeHi48dvDgQXz77bcV+vxz585Fdna24vFBgwbJYsXFxUYtUf3777/j8uXLsvjAgQNV56wpli1bVub7VZYFCxYIbygo7xquV68eevbsKYv/9ttvRs1SVzq3ReecpXv99deFeyWHh4ejoKDADBVZrrp168pimZmZZqiEiIiIiCwJG8BERERERFRh6tati1GjRsniqampCA0NNUNFppWdnY1Zs2YZPC41NRWff/65LF6vXj2MHDmyzLGNGjWSxS5evKiqCVxSUoIpU6aYfVasqQ0cOBBDhw4VHpsxYwb2799v8ufMysrCW2+9hYULF5b5egYHB8PJyUkWX7x4saqbCYqLi/Hee+8Jj73zzjsG56tpbt68qXjDQFliYmKwZs0aWbxFixbo3bt3ueNF701RURFmzJhhcC0AcPLkSeEs8A4dOsDf319VTnOqXbs2pkyZIounpqbio48+MkNFlku03PilS5fMUAkRERERWRI2gImIiIiIqELNmTNHuKTxihUrhA2Uqmbz5s1YtGiR3o9/8OAB+vfvL5x1GBISIpzh+6yXXnpJFisuLsY333yjdw1PffTRRzhy5IjB46qCb7/9VtgYyc/Px6BBg4xebvdZhw8fho+Pj17LCdeuXRsTJkyQxbOzszFu3DgUFhYa9Nxz5sxBfHy8LP7yyy/Dx8fHoFw11eLFiw06H1JSUjBkyBCUlJTIjs2cORMajabcHEOHDhXuB71jxw6DPxczMzMxfvx44Y0HVfkmgFmzZgmXt1+yZIlJtxG4fPlyld5bWLTk/c8//2yGSoiIiIjIkrABTEREREREFcrT0xNz586VxSVJwsSJE7Fo0SIUFxcbnPfQoUPo16+fKUpUxdra+q//DgsLw4wZM5CXl1fmmLi4OPj7+yMhIUF27IUXXsC8efPKfd727dsLG0eLFi3Se2ZrcXEx3n//fXz22Wd6Pb4qcnFxwc6dO1G7dm3Zsby8PIwaNQqjRo3CzZs3VT/HxYsXMWrUKPj7+yMpKUnvcaGhoXBxcZHF9+/fj9GjR+u9f+eiRYuwePFiWVyr1eKLL77Qu56a6uk1LEkSxo0bp9dnUVRUFPz9/YXnTffu3YWzVpWeW7QKAABMmTIFkZGReuW5d+8eBg4ciMTERNmxDh06YNy4cXrlsUQNGjTA0qVLhccmTpyImTNnGnzDxLOOHj2KYcOGoU2bNrh7967qPObm6+sri8XExGDhwoXCmxSIiIiIqGZgA5iIiIiIiCrc7NmzERAQIItLkoSwsDB4e3tj27Zt5e4LnJqaimXLlqFLly4ICAgw696106dPh42NzV///uqrr9CmTRt88skniI2NRUZGBvLz85GSkoLdu3dj1KhR6Natm3C2pkajwXfffQedTqfXc4eEhMhiBQUF6N+/PxYsWKA4m624uBh79+7FSy+9VKqx4uDggObNm+v13FVJt27dsH37dmETGAA2bdqEZs2aYcKECThw4IBezaSMjAxs3LgR/fr1Q7t27VTNJG7YsCFWrVolPBYZGYnOnTtjz549is2buLg49O3bF2FhYcLj77//Pvz8/Ayuq6Z5dvn2wsLCvz6LvvzyS8THx+P+/fvIzc3FlStXEBkZicGDByMwMBCpqamyXLVq1cKqVaug1er/Z5a3334bw4YNk8WLioowcuRIjB07FlevXhWOLSgowKZNm9CuXTscP35cWM/69euFqy9UJePHj8eYMWOEx5YtW4aWLVvi66+/1msJ/NzcXBw7dgyhoaFo1qwZ/Pz8sH379irfJB05ciRq1aoli8+dOxceHh4IDg7G7Nmz8emnnyI8PLzUT3W+CYiIiIioprMu/yFERERERETGsba2xrZt2xAQECCc/ZqQkIDhw4ejdu3a8PPzg5eXF5ydnaHVanH//n2kpaXh9OnTqvZIrSje3t5YsmRJqf1Xk5OTMX/+fMyfP9+gXAsWLEDfvn31fvy0adMQERGB27dvl4oXFhZi3rx5WLx4Mfz8/NC2bVvUq1fvr9cwKipK2ChZsWIFIiIicOXKFYPqrgqCgoKwZ88eDB8+HBkZGbLj+fn5WLNmDdasWYM6deqgc+fOaNGiBVxdXaHT6VBcXIysrCykpKQgMTERly5dMsmeyW+88QbeeecdREREyI5dunQJ/fr1Q+PGjREQEAB3d3fY2tri5s2bOH36NM6fP6+Y19fXFwsWLDC6vpqgf//+yMnJwfLly/+KJSQkYNasWQbv7f2f//wH7du3N7iGlStX4syZM0hOTpYdW7duHdatW4du3bqhffv2aNy4MbKzs3Hjxg38/vvvuH//vmLeJUuWCJcGror+/e9/IzMzE7t375YdS0lJwbRp0zB9+nS0bdsWnTp1gpOTExwdHZGbm4uHDx8iMzMTFy5cwMWLF1WtNmHpGjZsiPHjx2PlypWyY7du3cLWrVsVx1pZWSneSEJEREREVRsbwEREREREVCmcnJwQFRWFQYMG4cSJE8LH5OXl6b2MsSWYPn067ty5o7iUqz4+/vhjzJkzx6AxdevWxY8//ojevXsjPz9fdjw7Oxt79+7F3r17y821YMECvPXWW8JGZHUREBCAP/74A6NHj0Z0dLTi457OEDx27Jiq53F2dsb8+fPL3cf5qa+//ho5OTmKe77eunVLr32Fn+rWrRt+/fVX4WxAElu6dCnu3buHDRs2qBpvZWWF7777DqNHj1Y1vmHDhvj999/Rp08fXLt2TfiYU6dO4dSpU3rnDA8Px7Rp01TVY4lsbGywfft2TJ06FatXrxY+RpIkJCQkCG8wqgmWLVuGEydOIC4uztylEBEREZGF4BLQRERERERUaRo2bIjo6GiEhoaW2kNXLUOWW60oixYtwpo1a1C/fn2Dxjk7O2Pbtm0GzxZ+qnv37tixYwccHR1VjbexsUFERIRwf+bqyMPDA1FRUVi/fj28vLxMmrtevXqYO3curly5gpCQEGg0Gr3GabVafP/99/jqq69Qp04do2qYPHkyoqOjVZ8PNZWVlRXWrVuHzz//3ODlkl944QUcPHgQkyZNMqqGZs2aISYmBgMHDjQqj5OTE3744QeDbyipCqytrbFq1SpERkbC3d3dpLl79OgBBwcHk+asbDqdDseOHcPUqVOr/LLfRERERGQa5v9rCRERERER1Si1atXC4sWLce7cObz99tuq/ljduHFjzJo1C4mJiRVQoeHGjRuHy5cvIywsDJ6enmU+tmnTpggPD0dSUpJw/09DBAUFITY2FkOHDtW76ajRaNCvXz+cPXsWU6dONer5q6LRo0fjypUr2LJlC/r27av6RgQrKyu8/vrr2LhxI27fvo0FCxbA3t5eVa7p06fj/PnzGDt2rMGzd1999VUcPnwYq1atUtzrmMqm0WjwwQcf4OLFi3j33Xfh7Oxc5uPbtWuHb775BomJiejZs6dJanBycsKuXbuwfft2+Pj4GDTW3t4e7733HhITEzF8+HCT1GOpRowYgcuXL+Orr75CixYtVOXQaDTo3Lkz5s6di8uXL+Po0aNo2LChiSutfDqdDhEREbh58yZWrlyJCRMmoEuXLnBzc0PdunUt4oYpIiIiIqo8GskUmxcREREREVWg5ORkrF27Vhbv1asXevXqVen1kGk9fPgQu3fvxqFDh3Du3DkkJyfjwYMHKCgogE6nQ926deHu7o42bdqgY8eO6NOnD7y9vfVueJqCh4eHbP/h9evXKy77eunSJcTHx+PmzZvIzs5G7dq14e7uDm9vb7Rq1apCakxMTMSPP/6IqKgoJCUl4d69e8jNzYWdnR1cXV3RunVr+Pv7Y/DgwcLGSWpqKnJyckrFGjVqBCcnpwqp11JkZmYiOjoaJ0+exPnz55GcnIzbt28jJycHBQUFqFu3LhwcHODo6AhPT0906dIF3bp1Q9euXStktm1mZiZ27NiB6OhoxMfHIzU1FVlZWSguLoa9vT3c3NzQunVr+Pn5YciQIWjWrJnJa6huioqKYGNjI4sfPnwYfn5+snhJSQnOnz+PhIQE3L17Fzk5OdDpdGjSpAl8fHxMPoNcJC4uDrt378bx48dx6dIl3LlzBzk5ObC2tkb9+vXxwgsvwNvbG4GBgRg4cKDey45XN2fPnsW+fftw6tQpJCUlIS0tDVlZWSgqKoKdnR3q1q0LJycnvPjii2jdujW8vb3Rq1evatHwJSIiIiIqCxvARERERERE5TC0AUxElsPQBjAREREREVFVx/VfiIiIiIiIiIiIiIiIiIiqCTaAiYiIiIiIiIiIiIiIiIiqCTaAiYiIiIiIiIiIiIiIiIiqCTaAiYiIiIiIiIiIiIiIiIiqCTaAiYiIiIiIiIiIiIiIiIiqCTaAiYiIiIiIiIiIiIiIiIiqCTaAiYiIiIiIiIiIiIiIiIiqCTaAiYiIiIiIiIiIiIiIiIiqCTaAiYiIiIiIiIiIiIiIiIiqCTaAiYiIiIiIiIiIiIiIiIiqCY0kSZK5iyAiIiIiIiIiIiIiIiIiIuNxBjARERERERERERERERERUTXBBjARERERERERERERERERUTXBBjARERERERERERERERERUTXBBjARERERERERERERERERUTXBBjARERERERERERERERERUTXBBjARERERERERERERERERUTXBBjARERERERERERERERERUTXBBjARERERERERERERERERUTXBBjARERERERERERERERERUTXBBjARERERERERERERERERUTXBBjARERERERERERERERERUTXBBjARERERERERERERERERUTXBBjARERERERERERERERERUTXx/wAk/bTiWspfTQAAAABJRU5ErkJggg==)
This diagnostic graph displays the variance explained by each of the first 15 Principal Components (PCs 1-15) from Section 5.1. These metrics are valuable because they describe exactly how much variance in the dataset can be explained by each dimension in above 3D PCA. For example: if 75% of the genetic variance detected in your population can be explained by the first 3 principal components, you would have much more confidence in the relationships displayed in the PCA plot than if the first 3 PCs could only explain 25% of the genetic variance. Although no clear cutoffs exist, high levels of dataset variance (>50%) explained in the first 3 PCs inspire much greater confidence in the underlying relationships displayed than low levels of described variance.
5.3 Maximum Likelihood Phylogeny
![[RAxML Phylogenetic Tree]](data:image/png;base64,iVBORw0KGgoAAAANSUhEUgAABjkAABqvCAYAAABsPH8YAAAABHNCSVQICAgIfAhkiAAAAAlwSFlzAAA9hAAAPYQB1ayvdAAAIABJREFUeJzs3V2MG2ee7/dfsUl2r9iyWm9jsuWo1zMj2zOKEKlH9hlgR2vh7Gyc3AwQqY14rAsDR0CAXBz4KrnYLGAJcALk0giwe84iSF9oI89YRwYCJPCFZ5BRFvYGM9LxhQ0d7EyUY53IZNsta1riSxdZRVYuKLZbbZL1zqpifz8AAZqsp56nnnrYtJ4/n+dvOI7jCAAAAAAAAAAAIGNySTcAAAAAAAAAAAAgCIIcAAAAAAAAAAAgkwhyAAAAAAAAAACATCLIAQAAAAAAAAAAMokgBwAAAAAAAAAAyCSCHAAAAAAAAAAAIJMIcgAAAAAAAAAAgEwiyAEAAAAAAAAAADIpn3QDgEkyDONbrzmOk0BLAGzHZxMAAAAAAABBsJIDAAAAAAAAAABk0tCVHMN+UevHJH59O82/+vXS/2GuNcrzT/N9AIBpxncNAAAAAACYBrFsV7V9MiKOSYhREyeGYTDpASSIiUgA4G8hAAAAAACTFPt2VYZhhF4Zgm8L2qfcCwCAV3zXAAAAAACAtJtYTo6oJjzczsPECgAAAAAAAAAAu4Pn7arC7JsNAMA4bOUDAAAAAACAICJfyeE4zsjJKgIg0fLbn/Q/AMAvvmsAAAAAAECaxbZd1SQTjgc9Lkvi+pUzv54GAAzwXQMAAAAAALJmYjk5BqIOQDBxAgAAAAAAAADA7uQ5J0cQjuNMLOH4sOOnLQAyrD+9Xuew/pu2/onSqPEWZ5+5jfEo6/b6eZrWMZLU9aflHk/qvibxORplUv2Rhn4Pi++abIri8zbJv1FpqhsAAAAAkG2xBjkmZVLBlKgmCviHenq5jaPB+0kEHKKoO8ze+kFz7XipM+lJdy/HB22jnzrD1hdkS7+o6tl+niB/j4NOjkfRlkl9tobVQ1BgesT1tzCOz5uXOsYdl9XvQAAAAADAdJr4dlVRiuMfunH/4zmJXCXTmKMkDn4no8P2a9BzBCkXVXuzKonrD1vnpJI9R31f0zRO0tqHaeojP/iuSZ8sfQ+lqW4AAAAAwHTJRJBj0pN3QX+x7nZcFAGOqIIk/BLySZMeY0n88na3SuL6J11nWgJYaRpraQ9YpKmvhuG7Jv3S8LnPwncgAAAAAGD6ZXa7qp0TJ1FuWTXufG77kqdpD3p442W7mCjHVpCtzEaVCZt7xq3stE8ixXH9YfaVj7o+r1vfDF6PersmP/VHKWxbwuSfGFfnsDwXQBBRjPFR4v4eStN3IAAAAABgOsQa5JjWCZxR/6hO8nrT2KYsGjVZMng9SOBr57F+6vVSfxz1DTvOy7nSll9gEtfvpT6vdUZV37i6ohrL43ipf1Lc2hLH38ggf0eygu+a0ZL6W+j38zbp76G01A0AAAAAmF4T367K7z/yk9yOIa3bdYQ9H796HM7vJPQk6x13rN8xHvQapmXcJHH9QcrG3c4kJlwnLc7PdJgAWpr6aBy+a9Ivif9PiTrIlWTdAAAAAIDpEFuQI85/dLr9SnYSdXnddoRJnmyYxCRL2lY5IFqT/hsQ19/YNASGoxC2LUH6IezfEcCPIGMoye8hvgMBAAAAAHGJPMhhGEZkk31pSYbrFuhIS4CD/d53B37Jmh1p2popiuOnFf3gD981SPJ7iO9AAAAAAMBOnnNyxBUoiPo8ce2xPkpS/7AOep1M5qUH9wKIR9yfrd00ocp3zXRL8j4xRgAAAAAAUZlITo5p+IdsVn4h7ba6BN8WVb6EcX2epvuRprYkIY7rn/Q2LFHWF9Wvoqfh73wYk8y7kgZ81yQrqq2qJoVxAgAAAACIk+eVHEGEmcCJ+h/EhmFEkkTVS7smNXHl9xe2WZ5Qm0aTmPQZNUYGr037mNjt1w9Ege+a6UXgAwAAAAAwDWINckQ9kej1PHFuWeV2biZ3kCU7x3Iaxu8kVxGk8foBAAAAAAAAeOc5yOFl8m/U5GQUqyjgHb+OxHaDz56XcTGNk/67/fqBuPBdAwAAAAAA0iDSnBzjJgT9TIbENXES94RMEhOifla3YHcLuof7tExk7vbrB8LguwYAAAAAAKRV5NtVxblVVBomG92SSzPBA6/SEBTz+pmalrG9268fALZL8u8af1MBAAAAAFGJJSfHuGS/bv+ojTuQEfdkJZOhyBI/k/6TGtuT/Pyk8foBAAAAAAAAeBfpdlXbTeNkoJ9ffU+SW19P471APBzH2dXjZbdfPzAO3zUAAAAAACCNYgtyjJKGLaeC8NvurF4nJifNY2TUZGWa2xyl3X798C/I2GA8IWlJjkHGPwAAAAAgKrEGOfz+qnPUP3gHv64O8vBTj992DdqWBqPakZb2pVlUk5Pj+jqL9yGLbY6Sn+sfdmycE3hR1ud3LGN39w/fNdlF/g0AAAAAwLSa+EoOyd9kXNh/GMf5D+vBufnVNwAAAAAAAAAAkxd7kMNrAGDSAYGw+TV2XlcaAh1eV7IgPQiETbe0/l0LejzG89OfWe57vmumC1tWAQAAAACyLpGVHF5FNXES9DxeAxxBz4P0CDs56WVMxBUIG1d+t4+9SQcZJ9mGuCaWmbD2Jsz93u2fSyQnie+hNNQNAAAAAJhuEwlypGGVg19B2sbkYLYlOTkZNC+I1zZPKinypHNTeDHJ64/yXFHm2Ijy/HBH4BFp/FvoJs7voTTXDQAAAADIvnzSDZCS+4e/YRi+AxNuxzuOM/R6gtSVVVHdzyT6a9R9iir5/Kjxsb2OcecL07fbywatI+pVUWHPG7QNcV3/pO/vuL83o+qK4/7uVl7ud9DycJfF75o0/C1M8nsoyboBAAAAANNrYkEOt8m4YcdPov5hwraJQEe27LxfcU+iuI3FSUzixLlSKQsTt0lef9R9E3aifft54F/Q8U5/T780/y1M8nsoDd+BAAAAAIDpMtGcHFmY1Ik76MI/3tMp6P2ddLm4zxVVHVn4rPuR9usPW9+03a9JS/v4QHLSfK+T/B7K2ncgAAAAACDdUp14fBLi+gU/2y1kj99tp6KYWA5zDi/lo5j8CRPISXryKcvXP6lJw6Tv0bTw+nmkv3efNN/3SXwPpbFuAAAAAMB0MRz+hQh8y6S2UfNab5T1e80VMK2SvP5J7sdP7o1sGHafuD9IUpJ5Q9KQswQAAAAAkD0EOQAASAABDgAAAAAAgPB2/XZVAAAAAAAAAAAgmwhyAAAwYeRmAgAAAAAAiAZBDgAAAgoSrEgq5w8AAAAAAMA0yifdAAAAsmx70GJcoILVGwAAAAAAANEjyAEAQESCBjJYxQEAAAAAABAM21UBAJAgAhwAAAAAAADBEeQAACCgsAEKAhwAAAAAAADhsF0VAAAhbA9UeNmuisAGAAAAAABAdAhyAAAQEQIYAAAAAAAAk8V2VQAAAAAAAAAAIJMIcgAAAAAAAAAAgEwiyAEAAAAAAAAAADKJIAcAAAAAAAAAAMgkghwAAAAAAAAAACCTCHIAAAAAAAAAAIBMIsgBAAAAAAAAAAAyiSAHAAAAAAAAAADIJIIcAAAAAAAAAAAgkwhyAAAAAAAAAACATCLIAQAAAAAAAAAAMokgBwAAAAAAAAAAyKR80g2YFoZhbD13HCfUcYZhPPGeWxmvdQMAAAAAAAAAME1YyRGBQZBhEGDYHnTwe9yw1xzHcQ2IDB6j6gYAAAAAAAAAYNqwkiMigyCEW6Bh3HGDgEXcgYrarZqqt6qSpMXTi6osV4Yet/ROsPPffdO97N03/Z1z+/n8lPVbbli7/bbVz7n9nD9o23aWm3R9cdYZtk9HncutvN8+Dft5mNS9iHqMBm2jl7JR/40BAAAAAABANhHkSImdW1TF4drKNd2+fluFPQUZOUNy+sEWq2Xp+KvHtfLeSqz1b7f0TriJ7zjKjTreT1uDtGXwXpDJ53Fti6Pf3PoiaB8GqTNsn3o9V5DjohR1n8bRn3GNtXF1AgAAAAAAABJBjlQIE+DwsvLj3sf3tPryqkqHSjp65qiK80UZOUM9q6dOoyNzw9SdD+/o7dm3dfGji6qc7q/scJvQ9DL5OG4yNYrgQVRG/Yo/yra69aeXyecgbRu853cieuc5427nsDp3ntfL8X7rDCKqPnUTtE+Dlgvbn1H2i986AQAAAAAAsDuRkyMlDMPYegz+26vtOTl2uvfxPV392VVVlit69qfP6shLR7R4elHlk2Ud/sFhHTh2QAvPLujgcwdVerqkK69c0Re//SKy6xomyHZTQSeH/ZbbeXxUE6hRTNL6bdvdN6Ptt7ja6Xav/L7upU4/9e88bxKT6kHHpZ9yYfozaL+MK0fwAgAAAAAAAF4Q5IiI1+DEsOO2Bym25+wI0oad5VZfXtWB7x1QZbmixdOLeubHz6jyo4oO//Cw9n9/vxaWFvTUkac0X57X3speFeYKWj2z6rvuOAWd7PS7VZHbZGuSWxWlrW0760trO71K44R60D7N+r0AAAAAAAAA/GC7qggMtowaBC62Bxq2Bx7GHTfO9oDIzrI7gyXbXVu5ptLhkg4+d1CHXjik8nJZC0sL6na6an7VVH4uL8Mw1LN76na6sk1bVsuS+cjU+6+9r3O/OOe3KyI1qVwcSE7SeReSDg6lMbiSNfQpAAAAAADA7kaQIyKjAhY7X/cS2PBTZtx7t6/f1tEzR1V6uqS9R/ZqYWlB+47uk7lhqtvpympZ6tQ7Ks4X+49SUYVSQXP75/Tpe5/GFuTwMrE86W2qkkRQ5htRTVjHOcailIY2eJHGVUxRlwMAAAAAAEA2EeSYUrVbNRX2FFScL6qwp6BcIadupytzw5Rt2nJ6j1eVzBjK5XPKFXLKFXPKF/P9x1xea5+sqXyqHKodQZJGe30/6nJJS0O7777Zv2fDkj3HOdE96txhJ6zjGmNBTLI/4xJnfw3rD7/3L4t9CgAAAAAAgHDIyRFSrVZ74hHm9d/97ndb/33jxo1Qz6u3qjJyhoycIafnyGpaan7VVL1aV+t+S51GR7Zpq2f35PQcyXm89VVOMmb6Zao3qxH0kH+7aZuqNP/qfBDsGDwmYZCIOkyCby99Oumx4pbIPc1jN8kx6jdImpU+BQAAAAAAQHRYyRHC5cuXdezYMZ09e/Zb73ndvmr7671eb+hxgZ87Us/qyWpY2nywqfxcXlbLkmEYsk1b7UdtWU1Ltmmra3X7AY+u0w96RGTUxKjbaoHdsEVNWts8KjF13AmrR01ajxorw/gJcKSh3wfXl1aT7Kthdfi599vPk+Y+BQAAAAAAQLQIcoT0+9//Xq+//vq3Xl9cXBx6/LjXt7+3PXAS5HntqZocx1Gn0dHmxqZm12dlGIY69Y6MmX6ycatpqf2orU6zI6v1ZLDDMAwtvji8rVFwm7weN0k5buI1aLlJS1NbRklz24bx26dZGStJSUMf+A1yAQAAAAAAYPchyDGlKssVWS1L5oap1v2W8rN59eyeivNF5fI5OT1Htmmr0+yo/bCtTuNxoKNlyzZt2W1b5ZPh8nFguDRMHgeV1rantV1ZRX8CAAAAAAAgKwhyTLHjrx7XnQ/v9AMbM/3E48X5onKFnORIXasrq2Wp0+gHOgarOsyHpk68diKRNgfdZijM9kReV5VEMeHr91yTbFsYQdsZxdZCQfo0qnOF5ZZzIkyfBh0zaRlTQbFVFQAAAAAAwO5C4vEptvLeiqyWpXqtrsZaQ/VqXfUv6qpX+//d/LKp1npLm19vavPBptoP2zIfmlJPOnf1XKxtS+tE5M52RdnOsJPHcbbNy3m9tj9oO4cdNw0T8m79KU2uT93KpSnIM+79KPoUAAAAAAAA08FwRmXChqvLly/LcRxdunQp6aaMVLtZ05VXrqgwV9DcgTkVSgXli3kpJzldR12rK7tlb63gcHqO3vjVGyovj96qyk9yZzd+JiLjTkru9qv6MML2R5C2Ba3TrVzQ+x5mvPjNv+K13lHnjLJPvZSZdJ9G3Z9xjbUw5QhyAAAAAAAA7A6s5JhyldMVXfjggpr3m9r4fKO/mqNaV6PW2FrN0Vxvql6ry2pYrgGOqNx9M32TkOO2s0raJNs2ri63+oK2M0ydaTeu/Un1aVp4uQa/29NNw5gBAAAAAACAd6zkCCELKzm2u/7z6/rsl58pP5eX0+vfdsMwZLdtnXjtROxbVAEAAAAAAAAAECUSj+8i5989r/PvntfaJ2uq3qxKkhZfXFT5ZPwrNwAAAAAAAAAAiBpBjl2ofKqs8ikCGwAAAAAAAACAbJtokMMwjK3n43bJGnXcuPJB3wOC8JowWSI/AAAAAAAAAADEZWKJxweBhkGQYXvgwetxjuOMDFIEfQ8AAAAAAAAAAGTTRFdyDAINjuOMDHL4OS5OtVs1VW89zltxelGV5Uoi7UA6sToDAAAAAAAAAJJHTo4drq1c0+3rt1XYU5CRMySnH2yxWpaOv3pcK++tJN1EAAAAAAAAAAAgghxb7n18T6svr6p0qKSjZ46qOF+UkTPUs3rqNDoyN0zd+fCO3p59Wxc/uqjKaVZ2AAAAAAAAAACQJIIc6gc4rv7sqirLFR187qBKT5dU2FOQ03NkNSxtbmyqdb+l4nxR9VpdV165ogsfXEi62QAAAAAAAAAA7GoTDXIYhuEpz4bX46Ky+vKqKssVVZYrOvTCIe09sle5Qk5W09Lmg03Nrs8qP5tXbqafp71+r67VM6vSX02keQAAAAAAAAAAYIiJBTkGQYtB4GKQXFz6Jqjh5bidzwfvB33v2so1lQ6XdPC5gzr0wiGVl8taWFpQt9NV86um8nN5GYahnt1Tt9OVbdqyWpbMR6a617oSKToAAAAAAAAAAEjERFdybA9YjHvd63FRvHf7+m0dPXNUpadL2ntkrxaWFrTv6D6ZG6a6na6slqVOvaPifLH/KBVVKBU0t39OndudkecFAAAAAAAAAADxyiXdgCTVbtVU2FNQcb6owp6CcoWcup2uzA1TtmnL6T1eVTJjKJfPKVfIKVfMKV/MK1/MS3nJWJvMlloAAAAAAAAAAOBJEwly1Gq1Jx6S9I//+I9b79+4cSOR57959zcycoaMnNFPMt601PyqqXq1rtb9ljqNjmzTVs/uyek5kvN4u6uc+oEPIydV/fcHAAAAAAAAAAAIL/btqi5fvqxjx47p7NmzT7y+fQuppJ73X5B6Vk9Wo59kPD+Xl9WyZBiGbNNW+1FbVtOSbdrqWt1+wKPr9IMeAAAAAAAAAAAgMYYzLmFFBC5fvizHcXTp0qU4qwmk9m9rWj2zqvLJsg4cO6CFpQWVvlNScb4oY6afbNxqWmo/amvzj5tq3W+ptd5Sc72p5pdNPfzioXr/oqdL/+pS0pcCAAAAAAAAAMCuM9HE42lTWa7IalkyN0y17reUn82rZ/dUnC8ql8/J6TmyTVudZkfth211Gh1ZLUt2y+7n7LAdqZz0VQAAAAAAAAAAsDvt6iCHJB1/9bjufHinH9iY6SceL84XlSvkJEfqWl1ZLUudRj/Q0X7UVqfZkfnQlI4n3XoAAAAAAAAAAHavXR/kWHlvRW/Pvq16rS5Jsk1bxVJRuWJOhtHfsso2bVmt/rZV7YftfoCjJ2lFEqk5AAAAAAAAAABIRC7pBqTBxY8uympaqt+rq16t61H1kRrVhuq1uhprDTXXm2qtt7T5YFOtBy05PUcXP76YdLMBAAAAAAAAANjVCHJIqpyu6MIHF9S839TG5xuqV/vBjkat0Q9yfNlUc72peq0uq2HpjV+9ofIyyTgAAAAAAAAAAEjSrt+uauDIS0f01+2/1vWfX9dnv/xM+bm8nF5/LyrDMGS3bZ147YTOXT2XcEsBAAAAAAAAAIBEkONbzr97XuffPa+1T9ZUvVmVJC2+uKjySVZuAAAAAAAAAACQJgQ5RiifKqt8isAGAAAAAAAAAABpRZAjIoZhbD13HCfQcaPe2/662/vj6gYAAAAAAAAAYJqQeDwCgyDDIMAwLCjhdpxhGHIcZ+uxM3Cx/bHznKPKAQAAAAAAAAAwzVjJEZFB8MEt0DDqOK8rMAZBDb/ltqvdqql663G+kdOLqixXhh639I7vU0uS7r7pXvbum/7Ouf18o8qOqtNLXWHKehW2jmHlg1zbpOuLs84o75uXMTaqXr/H7xSkfBz3IuoxOonPXpjxBgAAAAAAgOwjyJEiQbed8lru2so13b5+W4U9BRk5Q3L6x1stS8dfPa6V91aCNTyApXfCTWL6OWbwnt/giJeyXoWtY1T5cf0YNEjl1tao2xm0zijvm9e+CtqnYUTdp3H0Z1xjLWidUX1uAQAAAAAAkH4EOVJkZ56NUVtT+S137+N7Wn15VaVDJR09c1TF+aKMnKGe1VOn0ZG5YerOh3f09uzbuvjRRVVO91d2uE0ueplAHDeZ6ifQ4VXQ+tyuNYq2Bq1j1AoDr20bvOd3InrnOeNu57A6d57Xy/F+6wwiqj51E7RPg5YL259B+iVInW6rbuK+/wAAAAAAAEgPcnJMuXsf39PVn11VZbmiZ3/6rI68dESLpxdVPlnW4R8c1oFjB7Tw7IIOPndQpadLuvLKFX3x2y9ibZOfSUevAZW7b/oPYHh5P6oJ0ijq2HlcmD4ZZVx/x9VOt3sc5L7GMcYGxyQxae63T4OUC9OfQfsl7D0M+pkHAAAAAADA9CDIEZHBllFuib9HHRc0YbhbudWXV3XgewdUWa5o8fSinvnxM6r8qKLDPzys/d/fr4WlBT115CnNl+e1t7JXhbmCVs+sBmpLXHbzhKXXAMCkt1HaWV9a2+lVGsdY0D7N+r0AAAAAAAAA/GC7qggMkogPAg6jto8ad5xbIvJxW1WNKndt5ZpKh0s6+NxBHXrhkMrLZS0sLajb6ar5VVP5ubwMw1DP7qnb6co2bVktS+YjU++/9r7O/eJcmG4JjUnY6Zd07oSkg0NpDK4AAAAAAAAAWcJKjog4jrP12Pm6l+O8vOe37tvXb2v/9/er9HRJe4/s1cLSgvYd3ac9h/ZobmFOs0/Nqjhf/OZRKqpQKmhu/5w+fe9TP5fvi59E4lFMAoc51yQmwQnmfGPpnScfYc7j9ZgkAw1hr3NS0pRwXXJfjZKGewsAAAAAAIDJYCXHlKrdqqmwp6DifFGFPQXlCjl1O12ZG6Zs05bTe7yqZMZQLp9TrpBTrphTvpjvP+byWvtkTeVT5VDtCJI02uv7fuqMK2l4lNIwIXv3zW8m3scle45aXJPVcY2xICbZn3FJQz6S7a+PCxKl4fMEAAAAAACA+LGSI6Tnn39etVpt6zGw/TW31z/66KOt92/cuBHJ89+8+xsZOUNGzpDTc2Q1LTW/aqperat1v6VOoyPbtNWze3J6juQ8zu+Rk4yZfpnqzWrY7gkkjsnfIOecxK/B0/yL86hWVfgxSGAdJsG3lz6ddIDBLZF7mgMeSYzRNH8uAAAAAAAAkC6s5AjhrbfeUrVa9bW9lNuxUT3vvyD1rJ6shqXNB5vKz+VltSwZhiHbtNV+1JbVtGSbtrpWtx/w6Dr9oEdERk1Suq0WCDO5OazsqPpG2c0BjlFbAcWdsHpUIMDPvfMT4EhDvw+uL63SHOAYd5zfzzsAAAAAAACyiyBHSIuLi6Ff/8lPfrL1/OzZs9E8f/2sVv92VZ1GR5sbm5pdn5VhGOrUOzJm+snGraal9qO2Os2OrNaTwQ7DMLT44vBriILb5PW4id8gE69+Jst3c4BjuzS3bRi/fRr1GJs2WQ1wDF4n0AEAAAAAALA7EOSYUpXliqyWJXPDVOt+S/nZvHp2T8X5onL5nJyeI9u01Wl21H7YVqfxONDRsmWbtuy2rfLJcPk4sogAx3hpbXta25VVaQ5wAAAAAAAAANsR5Jhix189rjsf3ukHNmb6iceL80XlCjnJkbpWV1bLUqfRD3QMVnWYD02deO1EIm1OcpuhNAY4vK54SXpiOGg7o9iuKUifRnWusMZde9g+DTpmCHAAAAAAAAAgS0g8PsVW3luR1bJUr9XVWGuoXq2r/kVd9Wr/v5tfNtVab2nz601tPthU+2Fb5kNT6knnrp6LtW1R5yFwO5/X99MU4BhVftR/R2XUef1uJeR2Pi/1pnFC3i+3/pQm16dp+BxEWaeXvgUAAAAAAMB0M5xRGbIjcvnyZTmOo0uXLsVZDUao3azpyitXVJgraO7AnAqlgvLFvJSTnK6jrtWV3bK3VnA4PUdv/OoNlZdHb1XlJ7mzGz8Tm26JhoPWF0dbo67D7Rf/UdbpVi7ofQ8zXiZ137wGVfzU6aXMpPs0LZ+huOtMc/ALAAAAAAAA0WAlx5SrnK7owgcX1Lzf1MbnG/3VHNW6GrXG1mqO5npT9VpdVsNyDXBE5e6b0U5Aup0r6vombVyC5UnW5aWf/bweRZ1pN679SfXpNBjXd9MwbgAAAAAAAOANKzl2kes/v67PfvmZ8nN5Ob3+bTcMQ3bb1onXTsS+RRUAAAAAAAAAAFEi8fgucv7d8zr/7nmtfbKm6s2qJGnxxUWVT8a/cgMAAAAAAAAAgKgR5NiFyqfKKp8isAEAAAAAAAAAyDaCHEAAXhMmS+QGAAAAAAAAAIC4kHgcAAAAAAAAAABkEis5gABYnQEAAAAAAAAAyWMlBwAAAAAAAAAAyCSCHAAAAAAAAAAAIJMIcgAAAAAAAAAAgEwiyAEAAAAAAAAAADKJIAcAAAAAAAAAAMgkghwAAAAAAAAAACCTCHIAAAAAAAAAAIBMIsgBAAAAAAAAAAAyiSAHAAAAAAAAAADIJIIcAAAAAAAAAAAgkwhyAAAAAAAAAACATCLIAQAAAAAAAAAAMokgBwAAAAAAAAAAyCSCHAAAAAAAAAAAIJMIcgAAAAAAAAAAgEwiyAEAAAAAAAAAADKJIAcAAAAAAAAAAMgkghwAAAAAAAAAACCT8kk3AIibYRhbzx3H8X3c9tfHvbfz3OPKAQAAAAAAAADCYyUHptog0DAIMOwMPHg9znGcrcew10cZVQ4AAAAAAAAAEB4RFRTjAAAgAElEQVQrOTD1BgEGx3FGBjn8HBen2q2aqreqkqTF04uqLFeGHrf0TrDz333TvezdN0e/N6rsuDJezhGkziB1+63D67n9XtOocpOuL846oxgrw87lVt5vn4YdX5O6F1GP0Tg/s1HUCQAAAAAA4BVBDsADr1teBS13beWabl+/rcKegoycITn9462WpeOvHtfKeyvBGh7A0jvDJyPHTQYP3gsarBhV5yTEdV3jrilokCpMHwZpZ9A6w/ap13MFOS5KUfdpHP0Zpl+C1pvEvQAAAAAAALsXQQ7AxbBcG14CHV7K3fv4nlZfXlXpUElHzxxVcb4oI2eoZ/XUaXRkbpi68+EdvT37ti5+dFGV0/2VHW4Tml4mkMdNTo6bpA1azkt5v+Wi4tafXiaft7/vtT8G7/mdFN55zrjbOazOnef1crzfOoOIqk/dBO3ToOXC9qfffnFbPePnbwVBDwAAAAAAECdycgAJuffxPV392VVVlit69qfP6shLR7R4elHlk2Ud/sFhHTh2QAvPLujgcwdVerqkK69c0Re//SLWNrlNkvoJfGznFnxJchsbLxPDfs/hJWAQdFLdzwS423Fx3bco+tRL/TvPm8Q48tunQcqF6c+w/RL0HrM1FQAAAAAAmBSCHJh6gy2j3PJseD0uKqsvr+rA9w6oslzR4ulFPfPjZ1T5UUWHf3hY+7+/XwtLC3rqyFOaL89rb2WvCnMFrZ5ZnUjb4M5rAGDSv2LfWV9a2+lVGifLg/Zp1u8FAAAAAABAGrFdFabaIIn4IHCxfbuo7dtHuR2385zD3ttZdly5ayvXVDpc0sHnDurQC4dUXi5rYWlB3U5Xza+ays/lZRiGenZP3U5XtmnLalkyH5l6/7X3de4X54J3yoT5zcHgdp6BNE5+T5Oo7lvY+pOqj/EFAAAAAACQDQQ5MPVG5c/Y+brX46J47/b12zp65qhKT5e098heLSwtaN/RfTI3THU7XVktS516R8X5Yv9RKqpQKmhu/5w+fe/T2IIcYRNie5kYjmoyeRKT8Pyi/htR3zcvxyQZaEhDG7yIa4zefbN/brfcImnvHwAAAAAAMP0IcgATVrtVU2FPQcX5ogp7CsoVcup2ujI3TNmmLaf3eFXJjKFcPqdcIadcMad8Md9/zOW19smayqfKodoRJGn0uLJBymx/3U/egUkHH9IwkTtu0jnO/ghz38YJkug8LkmPryjElcR9MOYmVScAAAAAAIBf5OTA1KrVak88on79xo0bgZ5Xb1Vl5AwZOUNOz5HVtNT8qql6ta7W/ZY6jY5s01bP7snpOZLzeOurnGTM9MtUb1Yj6qXoeJ0YHiQl9pqc2C3RdlwT0mn+pfpg4nncBHTU/N63Ybz0aRoCWJMYX1FI8xgFAAAAAACYFFZyYCpdvnxZx44d09mzZ7/1nt9tqby87ve5HKln9WQ1LG0+2FR+Li+rZckwDNmmrfajtqymJdu01bW6/YBH1+kHPSIyamJ03BY148oGLee2Lc64c+22AMeoxNRxJ6yO4r75CXCkod/jHF9RmOR2bUE/7wAAAAAAAJNAkANT6/e//71ef/31b72+uLg49Hi/r28PoPh5vnh6UY7jqNPoaHNjU7PrszIMQ516R8ZMP9m41bTUftRWp9mR1Xoy2GEYhhZfHN6mKIQNOmR94jNNE+2jpLltw/jt03HBhSzcn7glHeAYvD4Nn3cAAAAAAJB9BDmACassV2S1LJkbplr3W8rP5tWzeyrOF5XL5+T0HNmmrU6zo/bDtjqNx4GOli3btGW3bZVPhsvHgeGyPIGe1rantV1ZRX8CAAAAAAA8iZwcQAKOv3q8n4NjvaXGWkP1ar3/qNXVWGuoud5U635Lm3/cVPthe2tVh/nQ1InXTiTdfF/imoyNeishv5PHbltEpWUyOmg7o2h3kD4d9dh5TNzGja+wfRp0zKRlTAEAAAAAAKQJQQ4gASvvrchqWVtBjXq1rvoX/UBHY62h5pdNtdZb2vx6U5sP+oEO86Ep9aRzV8/F2ja3CVi/5dyOcdv33+08SUzGj2vPsP+OStiAStB2+r1vftqUpCjGV1R96vXzNen+DPr3AAAAAAAAYFIMZ1RW5YhcvnxZjuPo0qVLcVYDPCEL4652s6Yrr1xRYa6guQNzKpQKyhfzUk5yuo66Vld2y95aweH0HL3xqzdUXh69VZWf5M5udp4jaDmv5f0EObzU51Wc1xW0L0aVD9KHXsqGGS9B75tbvaPOGWWfhh1fcfRp1P0Zd9lJ3X8AAAAAAIBRWMkBJKRyuqILH1xQ835TG59vbG1Z1ag1tlZzNNebqtfqshqWa4AjKqO2A3KbkHTbRmhcAuNx7wWtb1KCtD2OurzcHz+vR1Fn2oUdX3H0aZq4fTbT1l4AAAAAALA7sZIDUylr4+76z6/rs19+pvxcXk6v/5E0DEN229aJ107EvkUVAAAAAAAAAGRRPukGAJDOv3te5989r7VP1lS9WZUkLb64qPLJ+FduAAAAAAAAAEBWEeQAUqR8qqzyKQIbAAAAAAAAAOAFQQ5I6m+NNDBuB7NRx40r73Zur3UDXnhNeiyRUwAAAAAAAADIOhKPYyvIMAgwbA86eD3OcZyRAYpx7xmGsfW+4zgj6wYAAAAAAAAAYCdWckDSN4ELt0CD1+PiVLtVU/XW47wVpxdVWa4k0g6kE6szAAAAAAAAgN2DIAcStTNYMm67qmsr13T7+m0V9hRk5AzJ6R9vtSwdf/W4Vt5bmUSTAQAAAAAAAAApQZADiRpsVzXqvyXp3sf3tPryqkqHSjp65qiK80UZOUM9q6dOoyNzw9SdD+/o7dm3dfGji6qcZmUHAAAAAAAAAOwGBDmQavc+vqerP7uqynJFB587qNLTJRX2FOT0HFkNS5sbm2rdb6k4X1S9VteVV67owgcXkm42AAAAAAAAAGACCHJA0jcrKNzybHg9LiqrL6+qslxRZbmiQy8c0t4je5Ur5GQ1LW0+2NTs+qzys3nlZnKSpPq9ulbPrEp/NZHmAQAAAAAAAAASRJADW0GLQeBi1PZRbsftfD54f9x743JyXFu5ptLhkg4+d1CHXjik8nJZC0sL6na6an7VVH4uL8Mw1LN76na6sk1bVsuS+chU91pXIkUHAAAAAAAAAEw1ghyQNDrh987XvR7n9b1x79++fltHzxxV6emS9h7Zq4WlBe07uk/mhqlupyurZalT76g4X+w/SkUVSgXN7Z9T53ZnbJ0AAAAAAAAAgOzLJd0AYJjarZoKewoqzhdV2FNQrpBTt9OVuWHKNm05vcerSmYM5fI55Qo55Yo55Yt55Yt5KS8Za5PZUgsAAAAAAAAAkAyCHLtcrVZ74iFJf/jDH7bev3HjRiLPf/Pub2TkDBk5o59kvGmp+VVT9WpdrfstdRod2aatnt2T03Mk5/FWWDn1Ax9GTqr67w8AAAAAAAAAQHawXdUudvnyZR07dkxnz5594vXt20cl9bz/gtSzerIa/STj+bm8rJYlwzBkm7baj9qympZs01bX6vYDHl2nH/QAAAAAAAAAAEw9w3FLmBDS5cuX5TiOLl26FGc1CCDN96b2b2taPbOq8smyDhw7oIWlBZW+U1Jxvihjpp9s3Gpaaj9qa/OPm2rdb6m13lJzvanml009/OKhev+ip0v/6lLSlwIAAAAAAAAAiAkrOZBKleWKrJYlc8NU635L+dm8enZPxfmicvmcnJ4j27TVaXbUfthWp9GR1bJkt+x+zg7bkcpJXwUAAAAAAAAAIE4EOZBax189rjsf3ukHNmb6iceL80XlCjnJkbpWV1bLUqfRD3S0H7XVaXZkPjSl40m3HgAAAAAAAAAQN4IcSK2V91b09uzbqtfqkiTbtFUsFZUr5mQY/S2rbNOW1epvW9V+2O4HOHqSViSRmgMAAAAAAAAAplou6QYA41z86KKspqX6vbrq1boeVR+pUW2oXqursdZQc72p1npLmw821XrQktNzdPHji0k3GwAAAAAAAAAwAQQ5kGqV0xVd+OCCmveb2vh8Q/VqP9jRqDX6QY4vm2quN1Wv1WU1LL3xqzdUXiYZBwAAAAAAAADsBmxXhdQ78tIR/XX7r3X959f12S8/U34uL6fX34vKMAzZbVsnXjuhc1fPJdxSAAAAAAAAAMAkEeRAZpx/97zOv3tea5+sqXqzKklafHFR5ZOs3AAAAAAAAACA3YggBzKnfKqs8ikCGwAAAAAAAACw2xHkwNQzDGPrueM4gY4L8t72193qBgAAAAAAAAD4R+JxTLVBoGEQYNgZePBynGEYchxn6+H1vcH5Bg8AAAAAAAAAQLRYyYGpNwgwDAtCeDluXIAi6uBF7VZN1VuP842cXlRluTL0uKV3gp3/7pvuZe++Ofq9UWXHlXErP4myQc7t5/xB27az3KTri7POsH066lxu5f32aZjPw6jycdyLqMdo0DZ6OUeU9x4AAAAAAMANQQ7AA69Bj6Dlrq1c0+3rt1XYU5CRMySnf7zVsnT81eNaeW8lWMMDWHpn+GTkuAnPwXt+Jz3H1eelvJeybsJc17jy49oWNEgVph+D9mGQOsP2qddzBTkuSlH36aQ/e3GI8t4DAAAAAAB4QZAD8GBnro1hAYudrw/L3bHztXsf39Pqy6sqHSrp6JmjKs4XZeQM9ayeOo2OzA1Tdz68o7dn39bFjy6qcrq/ssNtQtPLJOK4ydRxk7RByw0r66XcqF/xe63TC7e6vUw+B2nb4D2/E9F++zGKPvQ7+R60T8OKqk/dBO3ToOXC9mfQfhlXt98ycd97AAAAAACwO5GTA4jAqMDHOPc+vqerP7uqynJFz/70WR156YgWTy+qfLKswz84rAPHDmjh2QUdfO6gSk+XdOWVK/rit1/EdAV9bpOkfgIfA+MCL34mOnceG9UkqZ+gjNfjvAQMgk6qh+lHv+10C5oFGQ9+rttv0C6JifOg49JPuTD9Oel+Cfp3AgAAAAAAICiCHJh6gy2jxuXjGHecl3JBcnOsvryqA987oMpyRYunF/XMj59R5UcVHf7hYe3//n4tLC3oqSNPab48r72VvSrMFbR6ZtV3PVkx7BfmXifZk9yqKG1t21lfWtvpVRonxoP2adbvBQAAAAAAQBqxXRWm2iCJ+CBQMWrbqXHHjUtEPiwwMnh/Z3Bke7lrK9dUOlzSwecO6tALh1ReLmthaUHdTlfNr5rKz+VlGIZ6dk/dTle2actqWTIfmXr/tfd17hfnwnUM4CLp/AlJB4fSGFxJCn0DAAAAAADSjCAHpt6oVRY7Xx+3GsPrOby+d/v6bR09c1Slp0vae2SvFpYWtO/oPpkbprqdrqyWpU69o+J8sf8oFVUoFTS3f06fvvdpbEGOsAmxp23yk1/UfyOqiW4vfZqG8ZSGNniRxlVMUZcDAAAAAAAYhyAHMGG1WzUV9hRUnC+qsKegXCGnbqcrc8OUbdpyeo9XlcwYyuVzyhVyyhVzyhfz/cdcXmufrKl8qhyqHUGSRo8rO24LnqV3hicbzkoQIQ2Tskn146hzh52wDpLoPC5ZHZfbxdVfYfrGz98JAAAAAACAoMjJEZHBVkde8jcMO86tvNfz4xvPP/+8arXa1mNg+2vDXl9fX9eNGze2Xo/6efVWVUbOkJEz5PQcWU1Lza+aqlfrat1vqdPoyDZt9eyenJ4jOY+3vspJxky/TPVmNWz3RM7rL/S3P9Iszb86T6IfBwmlwySy9tKnkx4Xbonc0zxO4x6jcfRNmvsTAAAAAABkEys5IrA9j8MgEDFsq6Jxx43K4zB4bVQuCQz31ltvqVqtDu0nt62nHMf5Vk6OqJ/LkXpWT1bD0uaDTeXn8rJalgzDkG3aaj9qy2pask1bXavbD3h0nX7QIyKjJkZHrRZwKzuu3KiEymlNtJzWAEdS/TjqnnoZKwN+Ahxp6PfB9aVVkn3ltW/8/p0AAAAAAAAIgiBHRHYmsA57HMJbXFwM/Pp3vvOdrednz56N9Pni6UU5jqNOo6PNjU3Nrs/KMAx16h0ZM/1k41bTUvtRW51mR1bryWCHYRhafHH4NUTB7+S1n3JZmNhM00T7KGlu2zB++3TcBHoW7k/cstwHQf++AAAAAAAAjEKQIwN2BkTGreL4450/6nd/8zv9+//z3+vB//NAknTg2AE9e/ZZvfQvX9LCny7E3l6MV1muyGpZMjdMte63lJ/Nq2f3VJwvKpfPyek5sk1bnWZH7YdtdRqPAx0tW7Zpy27bKp8Ml48jTdI0YZumtviV1rantV1ZRX8CAAAAAAA8iZwcGTDYnmrwGLUC5MP/9kP93Yt/p0/+l09k/tHU/j/dr33/0T611lq69Xe39K9P/Wv9+q9+PeHWY5jjrx7v5+BYb6mx1lC9Wu8/anU11hpqrjfVut/S5h831X7Y3lrVYT40deK1E0k3f2LctmGKcsLX77km2bYwgrYzqT4d9dh5TNzGrSYJ26dBx0xaxlSat/ECAAAAAAC7D0GOKfH3f/n3+uR//kTlk2V9/z/7vr77F9/VkZeOqHyqrPKPyir/J2XNPz2v3/5Pv9XV//xq0s3d9VbeW5HVsraCGvVqXfUv+oGOxlpDzS+baq23tPn1pjYf9AMd5kNT6knnrp6LtW1uE7B+y3k5p5+cDl7b40fYyeM42+blvF7bH7Sdw47LyoT8OF7G+qT61Ovna1L9GbRvwvydAAAAAAAACMJwYs5gffnyZTmOo0uXLsVZTaJ2JhQfPA9y3LCk4m6Jx//+lb9X7ZOajv7kqA4eO6j5yrzyc3l1O121H7bV+rrVXzHwZUONtYYe3n2opZeX9IcX/zD19ybNajdruvLKFRXmCpo7MKdCqaB8MS/lJKfrqGt1ZbfsrRUcTs/RG796Q+Xl0VtV+Unu7GbnOYKW81LW70Syn7JuwlyXW/kgZcaVD9OPQfswSJ1h+3RcO6Ls06DJs72Uj7JcnJ+9UWXDJBYPUh8AAAAAAEBQrOSIwCDgMCxwMSyXxqjjBq9vfz44bvDazgDHv3v/3+ne/31PR146ou/8x9/R4ulFPfPPnlH5ZFkHnzuofUv79NSRp1R6uqTS4ZL2HNyj+cq8Pv/N59I/RdwR8KVyuqILH1xQ835TG59vbG1Z1ag1tlZzNNebqtfqshqWa4AjKqO2A3KblBy3jdC4171MdqY5kfkk2xamH4O2M+y9S7Nx7U+qT9MiaN+E+TsBAAAAAAAQBCs5Mu5vj/+tcvmcnvmzZ/TMP3tGiy8t6uBzB7X59eY3eR52Pr6o69G9R2qqKee/5t6kwfWfX9dnv/xM+bm8nN43wTC7bevEaydi36IKAAAAAAAAALIon3QDEFyj1tDXd77W0pkl/cn+P9HswqyKpaIkKZfPaWZ2Rvm5/Lcff5JXoVSQ/oNkNIcnMcdknX/3vM6/e15rn6yperMqSVp8cVHlk/Gv3AAAAAAAAACArCLIkWHVW1XN5Gc0MzsjY8ZQz+7J3DAlQ+rZPdmmrZ7dk5zHW2DlDOVmcsrl+w9jxpBTjXUhD3wqnyqrfIrABgAAAAAAAAB4QU6OEG7cuKFbt2498RgY/Pe1a9e2Xrt8+XKkzzf+341+oKLnqNvuJxlvrDVUr9bV+qql9kZbVtOSbdrqWl05Xae/FdLjoMeMMSM9iKgzgJRYesf7AwAAAAAAAEC2kZMjhKSv7ff/++/1b/7Lf6MjLx3RoRcOaeHZBc0/Pa/i3qJyMzl1O111Gh2ZG6ZaX7fUWm+pud5U88umGmsNbfx/G3JedfTW//pWIu0H4uAneEECZAAAAAAAACDb2K4qwxZ/tKhet6f2o7Y2H2yqsKcgp+eoWC8ql8/J6TqyNi116p3+o9mR3bL7Kzs6XTk9R84i21VhuhC4AAAAAAAAAHYPghwZNl+Z18HvH9Tm/U019zWVy+fUtboqzhc1U5jpb2PV6arT7Kjz6PGj0ZHVsmRtWtJBSaWkrwIAAAAAAAAAgGAIcmTcP/8f/rmuv35dhb0FGYYhu21vBTkkqWf1ZJmWOo2O2o/aMh+Z6jQ6sjdt6b9IuPEAAAAAAAAAAIRAkCPjnv/Z8/rTl/9Ud/+vu3J6/e2piqWiZoozkiE53f5qDqv1ONDxsK3W/Za+959+T/907J8kdqsCAAAAAAAAAGRULukGILzX/4/XdfQnR/XoPzxSo9pQvVpXvVpXo9ZQ48uGml821VxvqnW/peb9pr770+/qtf/ttaSbDQAAAAAAAABAKKzkmBIXPrigX/93v9bNv7mpxpcNzczOKD/bv722aavb7mrmT2b0Z//Nn+ns5bPJNhYAAAAAAAAAgAgQ5Jgif/Hf/4VO/1en9du/+a0+//Xn+voPX0uSDhw7oO/+xXf10r98SU8981TCrQQAAAAAAAAAIBoEOabMvqV9+sv/8S+TbgYAAAAAAAAAALEjJwcAAAAAAAAAAMgkghwAAAAAAAAAACCTCHIAAAAAAAAAAIBMIsgBAAAAAAAAAAAyiSAHAAAAAAAAAADIJIIcAAAAAAAAAAAgkwhyAAAAAAAAAACATCLIAQAAAAAAAAAAMokgBwAAAAAAAAAAyCSCHAAAAAAAAAAAIJMIcgAAAAAAAAAAgEzKJ90ApINhGFvPHcfxfdz214edw8v5DcMYWzcAAAAAAAAAANuxkgNbAYhBgGFnwMLrcY7jbD38nn9UnQAAAAAAAAAAjMJKDkj6JgDhOM7YgIPX4/yUG6zg8Hq+2q2aqreqkqTF04uqLFeGHrf0jufmPeHum+5l7745+r1RZYOU8XOOYedxO96PINflVt5L2aDXFFV9cdYZtk9HnSvqcRLm8zCqfBz3Iuox6uc+THLcAAAAAAAA4BsEORAZr1te7Szj9dhrK9d0+/ptFfYUZOQMyenXY7UsHX/1uFbeWwnU7iCW3hk+CTluMnjwXhyTl0EDOlGc38t1jSo/qh/d6hzHra1RtzNonVGOFa99Ffc48VNn1PciTH+G6Ze46nXrHwAAAAAAAPQR5EAkhm1R5Ra88BrguPfxPa2+vKrSoZKOnjmq4nxRRs5Qz+qp0+jI3DB158M7env2bV386KIqp/srO9wmFr1MII6bTB03Cem3nJfJXj/tjWMy260/vUw+b3/fSz9uL+P3mnaeM+52Dqtz53m9HO+3ziCi6lM3Qfs0aLmw/em3X9xWz/itd9S5AQAAAAAAMBo5OZAowzC2HoP/3u7ex/d09WdXVVmu6NmfPqsjLx3R4ulFlU+WdfgHh3Xg2AEtPLugg88dVOnpkq68ckVf/PaLWNvsNlnpJ/ARlXH1RnX+IO+NO85LwCDopLqfCXC347xuxeT3vkfRp17q33neJFYH+O3TIOXC9GfYfglyj4OOGwAAAAAAADyJIAckaWSQIehxXsptT1S+PWfHdqsvr+rA9w6oslzR4ulFPfPjZ1T5UUWHf3hY+7+/XwtLC3rqyFOaL89rb2WvCnMFrZ5Z9dW2NMv6r7m9TuRO+jp31pfWdnqVxgnxoH2a9XsBAAAAAACAyWK7Kmwl/R4EILYHGrZvKeV23M5zejn/ONdWrql0uKSDzx3UoRcOqbxc1sLSgrqdrppfNZWfy8swDPXsnrqdrmzTltWyZD4y9f5r7+vcL84F6I10SuMkNuLNs+Kn/qTqY1wGk/S4AQAAAAAAmCYEOSBpdOBh5+tej/P7/rBjbl+/raNnjqr0dEl7j+zVwtKC9h3dJ3PDVLfTldWy1Kl3VJwv9h+logqlgub2z+nT9z6NLcgRNiH2tE1s8ov6b0QVBPDSp2kYT2logxdxjdG7b/bP7ZZbxOuWZ9vPCwAAAAAAAG8IciCVardqKuwpqDhfVGFPQblCTt1OV+aGKdu05fQerw6ZMZTL55Qr5JQr5pQv5vuPubzWPllT+VQ5VDuCJI0eVzauXAtpkIZ2jpt0jjMYM+rcYe9hkETncZlkf8YlriTugzHnt864xg0AAAAAAMBuQpBjl3v++edVq9W2/rtSqUjSE68Fff0f/uEfdObMGUnSjRs39PLLL3t+/pt3fyMjZ8jIGXJ6jqympeZXTXU7XTk9R51GR7Zpq2f35PQcyXm8ZVZOMmb6Zao3q6GDHFEb9YvvrErzZGwSk/BRBAK89Omkr21UIvdBO9I8rtM8RgemIYAEAAAAAACQFIIcu9hbb72larU6dCspv9tSuZ3D7/P+C1LP6slqWNp8sKn8XF5Wy5JhGLJNW+1HbVlNS7Zpq2t1+wGPrtMPekRk1MTouC1qxpX1Ui4r0jp5PCoxddwJq8cFArzecz8BjjT0+/ZARxpNoq/G1RH074TfcQMAAAAAALCbEeTY5RYXF2N7/c///M+3np89e9bf89fPavVvV9VpdLS5sanZ9VkZhqFOvSNjpp9s3Gpaaj9qq9PsyGo9GewwDEOLLw5vaxSCTkJ6LZemiexh0t4+Kd1tG8Zvn44LLmTh/sQt6QDH4HWCFQAAAAAAAPEiyIFUqixXZLUsmRumWvdbys/m1bN7Ks4Xlcvn5PQc2aatTrOj9sO2Oo3HgY6WLdu0ZbdtlU+ma6uqaZHlCfS0tj2t7coq+hMAAAAAAGD3yCXdAGCU468eV71aV2u9pcZaQ/Vqvf+o1dVYa6i53lTrfkubf9xU+2F7a1WH+dDUiddOJN38wKZp+x+3LaLSMhkdtJ1RtDtIn4567DwmbuPGatg+DTpm0jKmxklz2wAAAAAAALKGIAdSa+W9FVktayuoUa/WVf+iH+horDXU/LKp1npLm19vavNBP9BhPjSlnnTu6rlY2+Y2Aeu33DBpmwgNO3m889rjCuaEDagEbeew46ZhQt7LWJ9Un3r9fE26P4P+PRh1TBbGBQAAAAAAQFoYzqhM0hG5fPmyHMfRpUuX4qwmEdN8bWlRu1nTlVeuqDBX0NyBORVKBeWLeSknOV1HXasru2VvreBweo7e+NUbKi+P3qrKT3JnNzvPEbTcsKcKC+EAACAASURBVHP4meCMot44z+/lF/9R1elWLuh9DzNeRiWl9iLIOIiyT72UmXSfRt2fcZcNej8IcgAAAAAAALhjJQdSrXK6ogsfXFDzflMbn29sbVnVqDW2VnM015uq1+qyGpZrgCMqo7YDcpuUdNtGaFp/wT0uMfMk6/Jyf/y8HkWdaTeu/Un1aZqM6wO3/pnmcQMAAAAAADAprOQIYZqvLY2u//y6PvvlZ8rP5eX0+sPWMAzZbVsnXjsR+xZVAAAAAAAAAIB0ySfdAMCr8++e1/l3z2vtkzVVb1YlSYsvLqp8Mv6VGwAAAAAAAACA9CHIgcwpnyqrfIrABgAAAAAAAADsdgQ5gF3Ia6JlidwAAAAAAAAAANKLxOMAAAAAAAAAACCTWMkB7EKszgAAAAAAAAAwDVjJAQAAAAAAAAAAMokgBwAAAAAAAAAAyCSCHAAAAAAAAAAAIJMIcgAAAAAAAAAAgEwiyAEAAAAAAAAAADKJIAcAAAAAAAAAAMgkghwAAAAAAAAAACCTCHIAAAAAAAAAAIBMIsgBAAAAAAAAAAAyiSAHAAAAAAAAAADIJIIcAAAAAAAAAAAgkwhyAAAAAAAAAACATCLIAQAAAAAAAAAAMokgB4D/n737j5HjzO/8/qma7p4Re0gOOeSqu6mdMbUi9wfNHDkiFeW8DAl7bSVA1oDJEcKVgJMh4vJXDkISJEDubrEioDsg+cfW5WLfXQwRxhLUrggKCJDAPshrL3O7vFgngggk8GDdySva1MysSEpD9q/qququ/NHs0bDVP6qqq7q7et4voLHNrnrqefpbNSvg++3neQAAAAAAAAAgkShyAAAAAAAAAACARKLIAQAAAAAAAAAAEokiBwAAAAAAAAAASCSKHAAAAAAAAAAAIJFSox4AgHgYhrHx3vO8gc4zDONLx3q189s3AAAAAAAAAAyCmRzABGoVGVoFhs1Fh6DndfvM87yN1+Zzeh0DAAAAAAAAgCgxkwOYUK3CRb9CQ6/zWgWLIIWKMDM3Vq+vauX6iiSpcKyg/FK+43mLrwe+tCTp1iv92956pfuxbm17tenV3k+7Qfv0I+rv5bdtmHhE2V+cfUZ53zZfq1/7oDEd5O+hW/s47sUo/vb69R/3MwcAAAAAAIKhyAGgo05LVEXt8vJl3bxyU+ltaRmmIXnNIolTcXTo+UNafms51v43W3y9cxKyVzK4daxb8jJsUWaQPofVR7f23eLYr89e+o016nGG7TPK++Y3VmFjOoioYzpOf3t++w7Tb7/4AAAAAACAcChyAPiSfgUOv7M7ul3n9rXbunDygrJ7slo4saDMbEaGaajhNGSXbFnrlj565yO9Nv2azv38nPLHmjM7+iUW/SQQeyVTeyUhw7bb3DZo4nWQPsNe308f3WYYDDsecY+zU5/t1/VzftA+w4jyGeslbEzDths0nmHj0qt/P9cK2w4AAAAAAATHnhwAOjIMY+PV+vdmm/fd6Na+W4Hj0m9fUn4pr/3f2a99z+xT4VhBuSM57f3mXu0+sFtz++c0f3Be2cez+uFzP9Qn734S/RfcpF+SNEjhw2/bsO2iSpD7SQwHvUYc8ehVwIprnP2KZmHuTZDvHbRoN4rZAUFjGqbdIPEcJC5h73/YdgAAAAAAYDAUOYAJ1a044ee8zQWMzXt2dGvffqzXTJALJy9o99d2K7+UV+FYQU88+4TyT+e191t7teupXZpbnNOOfTs0m5vV9vx2pWfSunDigr8vjdj5TeQO+1fr7f2N6zj9GseEeNiYJv1eAAAAAACA8cZyVcAEai0n1SpcbC44bC5A9Dqvl/aCSKdjnc65vHxZ2b1ZzR+c155v7FFuKae5xTnV7brKn5aVmknJMAw13Ibqdl2u5cqpOLIeWHr77Ns6/aPTQUMBBBLVnieD9j+q/saxuDJMYe//qJ8bAAAAAAC2MoocwITqVrBo/9xPYSNIm17Hbl65qYUTC8o+ntX2fds1tzinnQs7Za1bqtt1ORVHdtFWZjbTfGUzSmfTmtk1o/ffej+2IsegG2IPM7E5jD75Rf0XoioC+InpOCTKx2EMfgzrGQ17/ykeAQAAAAAwPBQ5AAzF6vVVpbellZnNKL0tLTNtqm7XZa1bci1XXuPhrJIpQ2bKlJk2ZWZMpTKp5msmpbUba8odzQ00jjCbRvdqG3fychR9DruffmNYfL3zBtNxJrq7XXvQIkCYjc7jMsx4xiWueIW9/3E9NwAAAAAAoDuKHMCEWV1dfeTf+Xw+ks9t29bi4qIk6erVqzp58mSg97N/NSvDNGSYhryGJ6fsqPxpWXW7Lq/hyS7Zci1XDbchr+FJ3sMlr0zJmGq2WXlvZeAiR9Q6Jd+T3Oc4J2NHkYSPohDgJ6bD/m7dNnJvjWMUz7Vfw3xGw97/SSggAQAAAACQFBQ5gAly/vx5HThwQKdOnfrSMb/LV3X7fPO/Q7/3pIbTkFNyVP2sqtRMSk7FkWEYci1XtQc1OWVHruWq7tSbBY+61yx6RKTXL7D7JXY7HfPTbhDD7HNcCxzdNqaOe8PqXoUAv/EPUuAYh7hvLnSMo1EWOFqf9bv/UTw3AAAAAADAP4ocwIT58MMP9cILL3zp80Kh0PH8oJ9LeqSI4vf96o5VeV5zxkZ1varpO9MyDEN20ZYx1dxs3Ck7qj2oyS7bciqPFjsMw1DhePcxDSpsEnIUycs4+hynRHs34zy2ToLGtFdxIQn3J27EAAAAAAAAdEKRA8BQ5JfyciqOrHVLlbsVpaZTargNZWYzMlOmvIYn13Jll23V7tdklx4WOiquXMuVW3OVOzJeS1VNiiQnj8d17OM6rqQingAAAAAAoBtz1AMAsHUcev6QiitFVe5UVForqbhSbL5WiyqtlVS+U1blbkXVz6uq3a9tzOqw7ls6fPbwqIc/kYImj/stETUuyeiw44xi3GFi2u3Vfk7ces0mGTSmg27mPaolquJsBwAAAAAABkeRA8DQLL+1LKfibBQ1iitFFT9pFjpKayWVf1lW5U5F1XtVVT9rFjqs+5bUkE5fOh3r2PolYIO2i2MsUfc5aPK4fRxx7eUwaEEl7Dg7nTdOCfmw/Dzrw4qp32d9FPEMc/8HaQcAAAAAAMJhuSoAQ3Xu5+f0w+d+qOLtopyKo3Q2rVQmJZmSV/dUd+pyK+7GDA6v4enctXOR9d8vqdpto+9+grTrtoF2FH36tfn6fn693/5Zq02ntv0KAP0+DxuPTuMIM85+7boZJKZhhY1p2IJUXDHtd8+DxnOQZy3s/Q/bDgAAAAAADIaZHACGKn8srxf/5EWV75a1/vH6xpJVpdXSxmyO8p2yiqtFOSVHL/3ZS8otxb8XR7flgPolpONYRmgUfQbVK5k9zL78xCrI51H0Oe56jX9UMR03Ye//JD83AAAAAACMK8PzPC/ODs6fPy/P8/Tqq6/G2c1ITPJ3QzIl7Zm88r0r+uDHHyg1k5LXaP5fkWEYcmuuDp89HPsSVQAAAAAAAACSjeWqAIzMmTfP6MybZ7R2Y00r761IkgrHC8odiX/mBgAAAAAAAIDko8gBYORyR3PKHaWwAQAAAAAAACAYihwAMAGCbpAMAAAAAAAATAI2HgcAAAAAAAAAAInETA4AmADMzgAAAAAAAMBWxEwOAAAAAAAAAACQSBQ5AAAAAAAAAABAIlHkAAAAAAAAAAAAiUSRAwAAAAAAAAAAJBJFDgAAAAAAAAAAkEgUOQAAAAAAAAAAQCJR5AAAAAAAAAAAAIlEkQMAAAAAAAAAACQSRQ4AAAAAAAAAAJBIFDkAAAAAAAAAAEAiUeQAAAAAAAAAAACJRJEDAAAAAAAAAAAkEkUOAAAAAAAAAACQSBQ5AAAAAAAAAABAIlHkAAAAAAAAAAAAiUSRAwAAAAAAAAAAJBJFDgAAAAAAAAAAkEgUOQAAAAAAAAAAQCKlRj0AAMDkMAxj473neaHOi+MYAAAAAAAAJhMzOQAAkWgVGVoFhs1FB7/nGYYhz/M2Xu3HwrQDAAAAAADA5GImBwAgMq0CRL9CQ7fz+s3ACNuuk9Xrq1q5viJJKhwrKL+U73je4uuBLy1JuvVK/7a3Xun8ud8+O7Xv1rZbX2EN2k+n9n7atrcbdn9x9Rn1fdt8vX7XCBrTsM91r/Zx3Iuon9EwfQXtO+zzDQAAAABbGUUOAMCWcnn5sm5euan0trQM05C8ZpHEqTg69PwhLb+1PLSxLL4ebRKzV6K1dSyK/gbtp1v7XvEIW2zqN9aoxxmmz6jvm99YhY3pIKKOabd2g8R0nOICAAAAAOiPIgcAYCy1lqCKqt3ta7d14eQFZfdktXBiQZnZjAzTUMNpyC7ZstYtffTOR3pt+jWd+/k55Y81Z3b0S4T6ST73SsJ2StL6SfZ2O6ffeKMqrITtp9sMA7/jax0LmhTuVliIa5yd+my/br9zg/YXVlQx7SdsTMO2GzSmQeIyyN9s2D4BAAAAAE3syQEAGDtxFDgu/fYl5Zfy2v+d/dr3zD4VjhWUO5LT3m/u1e4DuzW3f07zB+eVfTyrHz73Q33y7idRfJWu4khY+0nYjks/QQo7reNhk+pBEuD9zvO73FCQIlSU9y1oQn0UyyEFjWmYdoPEdBRxGdW9AAAAAIBJQJEDABCZ1j4Z/Tb+7nVerwJH2HYXTl7Q7q/tVn4pr8Kxgp549gnln85r77f2atdTuzS3OKcd+3ZoNjer7fntSs+kdeHEhZ7fYRSS/utuvwWAYX/P9v7GdZx+jWOyPGxMk34vxnVcAAAAADBJWK4KABCJ1mbgrQLE5oLD5gJEv/M2/+/m42HbXV6+rOzerOYPzmvPN/Yot5TT3OKc6nZd5U/LSs2kZBiGGm5Ddbsu13LlVBxZDyy9ffZtnf7R6YgiFJ1xTGIj2n1PBul/VP3xXHZHbAAAAAAgPhQ5AACR6TaTov1zv+eFvf5mN6/c1MKJBWUfz2r7vu2aW5zTzoWdstYt1e26nIoju2grM5tpvrIZpbNpzeya0ftvvR9bkWPUCemk95MEURQBgm4iPspk+jiMwQ+eUQAAAACYLBQ5AAATa/X6qtLb0srMZpTelpaZNlW367LWLbmWK6/xcHbIlCEzZcpMmzIzplKZVPM1k9LajTXljuYGGkevpKrfhHBUCeRhJaDHIdF965Vm3DptMB1norvbtQe5h2E3uI/LMOMZl7jjlZSiDwAAAAAkHXtyAAAGtrq6+sir0+c3btzY+Pzq1atDeb9yfUWGacgwDXkNT07ZUfnTsoorRVXuVmSXbLmWq4bbkNfwJO/hklemZEw126y8txI6LuNiWMnWcU7qtoodrdcwtDaTDruptN94DrvA0G/z9HEueIzzMwoAAAAACIeZHACAgZw/f14HDhzQqVOnvnTMz/JScb+XJzWchpySo+pnVaVmUnIqjgzDkGu5qj2oySk7ci1XdafeLHjUvWbRIyLdEqrdZhlEaasXOLptTB33htXdCgF+73nQAsc4xL31/cbVOMUKAAAAABAdihwAgIF9+OGHeuGFF770eaFQ6Ph+c0EkzveFYwV5nie7ZKu6XtX0nWkZhiG7aMuYam427pQd1R7UZJdtOZVHix2GYahw/ItxR81v0jtscnarFzg2G+extQsTz17FhSTcn7gNOwbEHAAAAACGhyIHAGBi5ZfyciqOrHVLlbsVpaZTargNZWYzMlOmvIYn13Jll23V7tdklx4WOiquXMuVW3OVOzLYfhyjQoGjv3Ec+ziOKemIKQAAAABMNvbkAABMtEPPH2ruwXGnotJaScWVYvO1WlRpraTynbIqdyuqfl5V7X5tY1aHdd/S4bOHRz38UMv/jGuBo98SUeOSjA47zkHHHeb7t+/70WkPkLB7ggTV61kdNKZhn5lRPFPjvGQXAAAAAEwiihwAgIm2/NaynIqzUdQorhRV/KRZ6CitlVT+ZVmVOxVV71VV/axZ6LDuW1JDOn3pdKxjC5IM9ZukHdcCR7f23f4dlUELKmHH2em8Xn2OS4Gnn37xlIYX037tRh3Tcb+XAAAAADApWK4KADDxzv38nH743A9VvF2UU3GUzqaVyqQkU/LqnupOXW7F3ZjB4TU8nbt2LrL++yVjo0qGbu7Hz6/qR9HP5s2pO7Xt94v8fp/3KiAEEXac/dp1Mqz71qvfXp+39xu2IBVXTPvd86AxHeRZC2sUfQIAAADApGAmBwBg4uWP5fXin7yo8t2y1j9e31iyqrRa2pjNUb5TVnG1KKfk6KU/e0m5pfj34ui3jNCof4kel17J7GH21a+/sOMcpM9x12v8o4rpOJnUv1kAAAAAGGeG53lenB2cP39enufp1VdfjbObkZjk74Zk4pnEKCTtubvyvSv64McfKDWTktdo/ifQMAy5NVeHzx6OfYkqAAAAAAAARIflqgAAW8qZN8/ozJtntHZjTSvvrUiSCscLyh2Jf+YGAAAAAAAAokWRAwCwJeWO5pQ7SmEDAAAAAAAgyShyAACwxQXZSJq9BgAAAAAAwDhh43EAAAAAAAAAAJBIzOQAAGCLY3YGAAAAAABIKmZyAAAAAAAAAACARKLIAQAAAAAAAAAAEokiBwAAAAAAAAAASCSKHAAAAAAAAAAAIJEocgAAAAAAAAAAgESiyAEAAAAAAAAAABKJIgcAAAAAAAAAAEgkihwAAAAAAAAAACCRKHIAAAAAAAAAAIBEosgBAAAAAAAAAAASiSIHAAAAAAAAAABIJIocAAAAAAAAAAAgkShyAAAAAAAAAACARKLIAQAAAAAAAAAAEokiBwAAAAAAAAAASCSKHAAAAAAAAAAAIJEocgAAAAAAAAAAgESiyAEAAAAAAAAAABIpNeoBAACA0TIMY+O953mBz+vXvtdxv30DAAAAAAB0wkwOAAC2sFaRoVVg2Fx08Hue53ldCxSGYWwc9zzvkXa9jgEAAAAAAPjBTA4AALa4VoGiX6HB73md2kRl9fqqVq6vSJIKxwrKL+U7nrf4erjr33qlf9tbr3Q/1q1trzb92ofpL2z/QfuJ63t1azfs/uLsc9CYdrtWv/ZBYzrI30O39nHci6if0bBjDHKNQZ43AAAAAF+gyAEAAEaivVjSqyByefmybl65qfS2tGRIeniqU3F06PlDWn5rOebRfmHx9c6JyF4Jz9axbgnMQdrGbdCxdWvfLY79+uyl31ijHmfYPqO8335jFTamg4g6psP+24tT2NgAAAAA+DKKHAAAYChay1P5/bck3b52WxdOXlB2T1YLJxaUmc3IMA01nIbski1r3dJH73yk16Zf08s/e1mF4wVJ/ROafpKIvZKpvRKRQdv1+xV+r7Z+EsVRJEz7xdNP8nnzcT9x3NwmaCK6W4zjGmenPtuv6+f8oH2GEVVM+wkb07DtBo1n2Lj06rubKJ43AAAAAF9gTw4AGCOGYWy8wp7X7djmz/30AUSpUwGjn9vXbuvSdy8pv5TX/u/s175n9qlwrKDckZz2fnOvdh/Yrbn9c5o/OK/s41ldfO6iPnn3k5i+QVO/JGmQwoffc0ad7PSTGA56DT8Fg7CJ4yAJ8H7n+V1uKOi9iyKmfvpvv+4onqWgMQ3TbpB4jiourb57/RsAAACAPxQ5AGBMRLEBdK+NnDd/HvU+CUi21nPip7jm57xO7cI8cxdOXtDup3Yrv5RX4VhBTzz7hPJP57X3W3u166ldmluc0459OzSbm9X2/HalH0vrjRNvBO5nEo1qCZ5OY+iX6B/2WNv7G9dx+jWOifGwMU36vfBjK3xHAAAAYNhYrgoAxsigG0D7TSQHSTrf+/CeJGn+4Lyv85EsrWeovXgmPfqc9Duv/X17Ea7Tc9rr+b28fFnZvVnNH5zXnm/sUW4pp7nFOdXtusqflpWaSckwDDXchup2Xa7lyqk4sh5Yevvs2zr9o9MRRCf5xjEBPinGea+UYfTHs/UFYgMAAACMFkUOAJgwYYoe7W78Hzf0b3/v3+ref7ynVKb5nwrXdjV/YF5/93/4uzry8pFIxorx0O05af/c73l+j/U6fvPKTS2cWFD28ay279uuucU57VzYKWvdUt2uy6k4sou2MrOZ5iubUTqb1syuGb3/1vuxFTkG3RC721JKi6/336NgHBOn/Nr8C1Eluv3EdByeiXEYgx+j3HB93GMDAAAATAqKHAAwYfpt5NxrFodbc/VH/+kf6f7f3tdjux7TvmP7ZGbMjU2ey3fK+tf//b/Wu//7u/r77/59GVPs64HorV5fVXpbWpnZjNLb0jLTpup2Xda6Jddy5TUeziqZMmSmTJlpU2bGVCqTar5mUlq7sabc0dxA4wizaXSvtv32DWgVOsL016nvYSZYxyGZ26tYFGeiu9u1B70PYTY6j8sw4xmXuOI1CbEBAAAAko49OQAAkqRasaZ/tv+fqbZe0+K3F/Xkbzypfc/uU+5ITl/51a9ozzf26Cvf+op2/spOff6Lz/V7C78nu2yPetgYwOrq6iOvlqtXr470/cr1FcmQDNOQ1/DklB2VPy2ruFJU5W5FdsmWa7lquA15DU/yHs5gMiVjqtlm5b2V4AGJ2aQlP8f51+qtYkevwlHUWhtYD7KRtZ+YDvs56reR+zg/13E/o0mODQAAADBJmMkBAGOkNcvCzwbQnc4Lu8GzJP3hr/6h0tm0nnj2Cc1/fV7bC9s1lZmSa7my1i1V7laav6rPmDIMQ/f/9r7+xeF/Ib0UqjuM2Pnz53XgwAGdOnXqS8c2P0Ojei9PajgNOSVH1c+qSs2k5FQcGYYh13JVe1CTU3bkWq7qTr1Z8Kh7zaJHRLolRnstLdWrbdglqfz0N2zjWuDotmlz3Js5h1mGrF2QAsc4xL31/cbVKGM17rEBAAAAJg1FDgAYE1FsAN1vI/JuRZA///6fq/agpie/86T2Htqr3N/JacdXd8hreKreq6r0aUlmymwmnlsbPddc3b91X/qppJNRRQHD9OGHH+qFF1740uebCx+jeF84VpAk2SVb1fWqpu9MyzAM2UVbxlRzs3Gn7Kj2oCa7bMupPFrsMExDheOFXl99IEGT1/3a9UvGBulvGIndcUq0dzPOY+skaEx7JdCTcH/iRgwAAACArYUiBwCMkUE3gA5zrOE29N4fvqf5g/MbGzzPf31eu57cpepnVRmm0SxqVFw55eZmz7ViTZnZjKZ3Tst515Hxn7M3B6KTX8rLqTgbM4hS0yk13IYysxmZKVNew5NrubLLtmr3a7JLDwsdFVeu1Xzljgy2Hwc6S3LyeFzHPq7jSiriCQAAAGw97MkBAFvc53/9udyKq5ldM5rePq30trQMw5BTcZobPJtGc2PntKmpzJSmpqeUmk4pNZ1S+rG05Eje59EtEQRI0qHnDzX34LhTUWmtpOJKsflaLaq0VlL5TlmVuxVVP6+qdr+2MavDum/p8NnDox7+SMS9PE7Q5HG/JaLGJRkddpxRjDtMTLu92s+JW6/nbdCYhn1mxuWZiiM2AAAAALqjyAEAY6BYLD7yCvP5rVu3No4Heb96fVUypan0lCTJrbmq3KuouFJU9V5VTtlR3a7La3jNmSAPN4Q2pgyZKVOmYUrjt8czEm75rWU5FWejqFFcKar4SbPQUVorqfzLsip3Kqreq6r6WbPQYd23pIZ0+tLpWMfWLzkZtN2g120XR3J00MRr+3eIqyAzaOI47Dg7nZeUhHwvfp7JYcXU79/PsOI5ytgAAAAAeBTLVQHAiP3xH/+x9u/fr1zui+V1tm/fLklaXV195Nxen//FX/yFfvd3f1eSAr2v3K3IMB/uc1B1ZH1uqbRW2tjk2ak2l6hyq67qdl0Np6FGvSE1mstfmYapRqURcVQA6eWfvayLz11U8XZRTsVROptWKpOSTMmre6o7zWXUWjM4PM/TuWvnIuu/X8Kx1wbiQdpt3qTYzy/Ah23zmIKOr99361cA6Pd52HvQaRxhxun33rUbJKZhhY1p2MR7XDHtd8+DxnPYz1rrWmFiAwAAAKAzihwAMGIff/yxfvGLX+jVV1/90rGDBw92bNPp81bxIuj7x/+Tx+XVveb+Bus1le+UZZiG7JItc8pU3a43jz2oySk/3ODZrqvu1OW5nuqNurzHWa4K0SscL+jFP31Rb5x4Q9YDSzO7ZpTKpGRMGfIanhpuQ67lbszgOHftnHJL8e/F4Sc5G6Td5mNhE57jPCugW1ziGOsgfYVtO8zvN2y9numtHtNRxQYAAADAl1HkAIAtLv90XnW7rtr9mir3Kko9lpLX8JQpPtzkud7c5LlWfLjvQWuTZ8uVW3Pl1T0Z+9h4HPHY98w+fb/2fV353hV98OMPlJppPp9Sc9k013J1+Oxh30tU+U0+hjVogjJs+zgTo1FcO+g1RnUPhhX/uAo8cfSZhGc6ac9LVO0BAAAANFHkAIAtLjOb0Vf/s6/q3n+4p+nt0zJTpupOXZlsRmbalDypbtflVJxmoeN+TXbRll22ZZdsaUHyUszkQLzOvHlGZ948o7Uba1p5r7kJTOF4Qbkj8c/cAAAAAAAA44siBwBAv3Pxd/TPD/5zlT4tSUZz8/FMNqOpTHMz8tayQK1lq1qFDtdypTMjHjy2lNzRnHJHKWwAAAAAAIAmihwAAO1c2KmTPzipf/NP/428hien6nxR5DAebvJs15ubkJceFjqKNf36P/l1vVN+R9oCEzkM44sluTyv+xfudt7mz9uPtx9rv77fvgE/gmyWzHI6AAAAAIBxR5EDACBJ+vb//G2lZlL683/057KLtqa3T2tqekqGacjzPDWch7M5irYcy9Fv/a+/peP/7XG9c/6dUQ89dq0ig+d5MgxDhmF0LDb0Oq9X4aLT8c3ntRdLKHQAAAAAAAA0UeQAAGx49r97Vgu/tqDL//VlldZKqrt1TaWbS1bV7eb77J6s/t6f/T3llrbWkkGbixWdZmUEOS9IoSJMQWP1+qpWrj/ct+JYQfmlHf7d1gAAIABJREFUfOBrYHIxOwMAAAAAMEkocgAAHlF4pqBX/voV/c3P/0a/+Mkv9LfX/laS9NVf+6r2//p+LXx7YcQjnExRLEl1efmybl65qfS2dHMGTqN5Hafi6NDzh7T81nIkYwUAAAAAABgXFDkAAF9mSAvfXqCgEYNOszg6LWXVbTmsTp/fvnZbF05eUHZPVgsnFpSZzcgwDTWchuySLWvd0kfvfKTXpl/Tyz97WYXjhWi/FAAAAAAAwIhQ5AAAIAF6FTgu/fYl5Zfymj84r+zjWaW3pZsbyJccVderqtytKDObUXG1qIvPXdSLf/riCL4BAAAAAABA9ChyAADgQ6vI0Gs/jiDnhem7kwsnLyi/lFd+Ka8939ij7fu2y0ybcsqOqp9VNX1nWqnplMwpU5JUvF3UGyfekP5hZMMDAAAAAAAYGYocAAD00SpatAoXmwsOmwsQvc5rP7f98/b+2o912rPj8vJlZfdmNX9wXnu+sUe5pZzmFudUt+sqf1pWaiYlwzDUcBuq23W5liun4sh6YMm+bEts0QEAAAAAABKOIgcAAD50m0nRb3+NMNfwe+zmlZtaOLGg7ONZbd+3XXOLc9q5sFPWuqW6XZdTcWQXbWVmM81XNqN0Nq2ZXTOyb9pdrwsAAAAAAJAU5qgHAAAAglu9vqr0trQysxmlt6Vlpk3V7bqsdUuu5cprPJxVMmXITJky06bMjKlUJqVUJiWlJGMtuiW1AAAAAAAARoGZHAAA9LC6uvrIv/P5/Eg+L5VKOnDggCTp6tWrmv2rWRmmIcM0mpuMlx2VPy2rbtflNTzZJVuu5arhNuQ1PMl7uOSVqWbhwzDVWGkECwYAAAAAAMCYocgBAEAX58+f14EDB3Tq1KkvHQu69NSgn2/+98Z7T2o4DTml5ibjqZmUnIojwzDkWq5qD2pyyo5cy1XdqTcLHnWvWfQAAAAAAACYABQ5AADo4cMPP9QLL7zwpc8LhULH8+P+XJJOnTql1R2r8rzmjI3qelXTd6ZlGIbsoi1jqrnZuFN2VHtQk1225VTaih2eJ69AsQMAAAAAACQbRQ4AABIov5SXU3FkrVuq3K0oNZ1Sw20oM5uRmTLlNTy5liu7bKt2vya79LDQUXGbe3a4npQb9bcAAAAAAAAYDEUOAAAS6tDzh/TROx81CxtTzY3HM7MZmWlT8qS6U5dTcWSXmoWO1qwO674lHRr16AEAAAAAAAZHkQMAgIRafmtZr02/puJqUZLkWq4y2YzMjCnDaC5Z5VqunEpz2ara/VqzwNGQtCyJ1aoAAAAAAEDCmaMeAAAACO/cz8/JKTsq3i6quFLUg5UHKq2UVFwtqrRWUvlOWZU7FVU/q6ryWUWe5+nctXOjHjYAAAAAAEAkKHIAAJBg+WN5vfgnL6p8t6z1j9dVXGkWO0qrpWaR45dlle+UVVwtyik5eumdl5RbYjMOAAAAAAAwGViuCgCAhNv3zD7949o/1pXvXdEHP/5AqZmUvEZzLSrDNORarg6fPazTl06PeKQAAAAAAADRosgBAMCEOPPmGZ1584zWbqxp5b0VSVLheEG5I8zcAAAAAAAAk4kiBwAAEyZ3NKfcUQobAAAAAABg8lHkAABgizMMY+O953mhzgtzbPPn7cd7HQMAAAAAAGhh43EAALawVjGhV3Gh33n9jnmet/FqL3hsfm3W6xgAAAAAAEALMzkAANjiWkWE9iJEkPP8XqObVjEk6LHNVq+vauX6w71IjhWUX8p3PG/x9cDDkyTdeqV/21uvdD/WrW2vNv3aD6NtmGsHuX7YsbW3G3Z/cfY5aEy7Xatf+6AxHeTvoVv7OO5F1M9o2DEGuUbY5xsAAADYiihyAACA2HSavRG1y8uXdfPKTaW3pSVD0sMunIqjQ88f0vJby5H32c3i652Tkb0Snq1j3ZKYo2rrx6DX79a+Wxz79dlLv7FGPc6wfUZ5z/zGKmxMBxF1TIf9txeXUfQJAAAAJB1FDgAAEJv2WRidZmWEncVx+9ptXTh5Qdk9WS2cWFBmNiPDNNRwGrJLtqx1Sx+985Fem35NL//sZRWOFyT1T2j6SSD3Sqb2StIGbdfvV/hxtQ2iXzz9JJ83H/c7rtaxoEnhbnGKa5yd+my/rp/zg/YZRlQx7SdsTMO2GzSeYePSq2+/7Sh6AAAAAP2xJwcAAEic29du69J3Lym/lNf+7+zXvmf2qXCsoNyRnPZ+c692H9ituf1zmj84r+zjWV187qI+efeTWMfUL0kapPDh95y424a9dpDrt5/np2AQNqkeJAHe7zy/yw0FjX8UMfXTf/t1R7EcUtCYhmk3SDxHEZdR3QsAAAAgyShyAACwxbWWk+q3l0av8/xeIyoXTl7Q7qd2K7+UV+FYQU88+4TyT+e191t7teupXZpbnNOOfTs0m5vV9vx2pR9L640TbwxlbOjPbwFg2L9ib+9vXMfp1zgmy8PGNOn3AgAAAEB8WK4KAIAtrLVnRqs40W1pqV7n+Tm2+d+bhVmq6vLyZWX3ZjV/cF57vrFHuaWc5hbnVLfrKn9aVmomJcMw1HAbqtt1uZYrp+LIemDp7bNv6/SPTgeKERBUFPudRNH/qPobx+LKqBAbAAAAIH7M5AAAYIvzPG/j1f65n/MGPdZrXJ3cvHJTu57apezjWW3ft11zi3PaubBT2/Zs08zcjKZ3TCszm/nilc0onU1rZteM3n/r/a79DWrQDbF7LaXUa7PlONpGgV/Uf2Hx9Udfg1zH7zmjTKYP+j2HZVQbiychNgAAAECSMJMDAAAkxur1VaW3pZWZzSi9LS0zbapu12WtW3ItV17j4aySKUNmypSZNmVmTKUyqeZrJqW1G2vKHc0NNI4wm0b3attv34BeidG42kZlHH65vjkOvTZoj1pcBaYwG53HZZjxjEtc8ZqE2AAAAABJwEwOAEBkWksW+dnbodN5vdpvPtar3bD2hJgEX//617W6urrxunr16saxcX2/cn1FMiTDNOQ1PDllR+VPyyquFFW5W5FdsuVarhpuQ17Dk7yH+4SYkjHVbLPy3oqf8AzVpCU/x2E2QTdRzaoIorWZ9CCbSvuJ6bCfo34buY/zcx33M5rk2AAAAABJw0wOAEAkNu/H0Co2dFpuqNd5rf/tVqgIs7QROvvBD36glZWVL+2fkYT38qSG05BTclT9rKrUTEpOxZFhGHItV7UHNTllR67lqu7UmwWPutcsekSkW2K022yBfm17teuVjO3X3yBtwxrXAke35bvi3rC6W7I7SPyDFDjGIe6t7zeuRhmrcY8NAAAAkEQUOQAAkWnfpHrQ8xCvQqHQ9d+nTp0ay/eFY80x2iVb1fWqpu9MyzAM2UVbxlRzs3Gn7Kj2oCa7bMupPFrsMExDheOPfu8oBU1e92vXLxnrZxmmMG3DGqdEezfjPLZOgsa0VwI9CfcnbsQAAAAAmDwUOQAAibG5INI+c6PXsc0+/+hz/bs/+Hf6xV/8Qp/9x88kSbsP7Nb+U/v1zD94RnO/MhfxqBGl/FJeTsWRtW6pcrei1HRKDbehzGxGZsqU1/DkWq7ssq3a/Zrs0sNCR8WVazVfuSOD7ceBzpKcPB7XsY/ruJKKeAIAAACTiT05AACJ4HneI6/2oka3Y5u98z+9o391/F/pxhs3ZH1uadev7NLOr+5UZa2i6//quv7l0X+pn/zDnwzrKyGkQ88fau7Bcaei0lpJxZVi87VaVGmtpPKdsip3K6p+XlXtfm1jVod139Lhs4dHPfyJFDR53G+JqHFJRocdZxTjDhPTbq/2c+LWazbJoDEN+8yMyzPFUlUAAABA9ChyAAC2hIu/eVE3/uiGckdyeuq/eEpP/saT2vfMPuWO5pR7Oqfc38lp9vFZvfu/vatL/+WlUQ8XPSy/tSyn4mwUNYorRRU/aRY6SmsllX9ZVuVORdV7VVU/axY6rPuW1JBOXzod69j6JWCDthv0uoO29WPQ5HH7OOJKAg9aUAk7zk7nJSUh34uf52pYMfX79zOseEYRGwAAAAD+sVwVACAyrU3E++2z4fe8qFx87qJW/79VLZ5a1PyBec3mZ5WaSalu11W7X1PlXkWV7RVNzUzJSBm69bNbuvRfXZKOD2V4COHln72si89dVPF2UU7FUTqbViqTkkzJq3uqO3W5FXdjBofneTp37Vxk/fdLqvbaQDxIu82bFPv5dXxUbf3afN2ox9evANDv87D3oNM4wozTb/zbDRLTsMLGNGxBKq6Y9rvnQeM57Gdt0D4BAACArYoiBwAgEq2iRatwsXlfjFZRw8957e9bx9sLIt3atR/792//e93+f29r4cSCvvKrX9Hjhx/Xjid2SIZkrVsqf1pWaiYlwzTkNTw1nIbqdl0f//RjaYekg2EjgjgVjhf04p++qDdOvCHrgaWZXTNKZVIyph7eR7ch13I3ZnCcu3ZOuaX49+Lwk5wN0m7zsSDJ4CjaDkO3uMQxtkH6Ctt2mN9v2Ho901s9poPGBgAAAEAwFDkAAJHptuF3++d+z4vi2E+//9Pm3hsLO7X7a7u191f3av7gvKr3qiquFOU1PNXtupyqI6fiyC7bsku2atmanJ84FDnG2L5n9un7te/ryveu6IMff6DUTEpe42FRzDTkWq4Onz3se4kqv4nZsAZNbo6y7zivG/Qao4pD2LbD/H5hrzms7zaKfif9eQEAAABAkQMAMMFKqyXd++ieFk8s6rFdj2l6blqZbEaSZKZMTU1PKTWT+vLrsZTS2bT0N5JRHs6SWgjvzJtndObNM1q7saaV91YkNWd65I7EP3MDAAAAAACMFkUOAMDEWrm+oqnUlKamp2RMGWq4DVnrlmRoYzmjhtuQvOaSV4ZpyJwyZaaaL2PKkLfSfZYIxkvuaE65oxQ2AAAAAADYSsxRDwAAkGynTp3Sd7/7XV2/fn3j1XL9+nX9/u///sa/z58/P9T363+9vrFHQ73W3GS8tFZScaWoyqcV1dZrcsqOXMtV3anLq3vN5Y4eFj2mjCnps1BhARJp8XX/LwAAAAAAxgEzOQAAAzl58mTXY08//bSefvrpjX//4Ac/GOr7D/+vD5szNqquag9qqtytyJwyZZdsmVOm6nZddqm5B4dT+aLY0XAbargN1Rt1abevMAAAAAAAAGAEKHIAACZW4emCGvWGag9qqn5WVXpbWl7DU6aYkZky5dU9OVVHdtFuvsq23IrbLHbYdXkNT16B5aqwdbDpMQAAAAAgaShyAAAm1mx+VvNPzat6t6ryzrLMlKm6U1dmNqOp9FRzGSu7Lrtsy37w8PVwVodTdaR5SdlRfwsAAAAAAAB0Q5EDADDRfv2f/rquvHBF6e1pGYYht+ZuFDkkqeE05FiO7JKt2oOarAeW7JItt+pKvzPiwQMAAAAAAKAnihwAgIn29d/+un7l5K/o1v9zS16juTxVJpvRVGZKMiSv3pzN4VQeFjruN/fu+NpvfU1/deCvJFarAgAAAAAAGFvmqAcAAEDcXvi/X9DCtxf04G8eqLRSUnGlqOJKUaXVkkq/LKn8y7LKd8qq3K2ofLesJ7/zpM7+n2dHPWwAAAAAAAD0wUwOAMCW8OKfvKif/KOf6L0/eE+lX5Y0NT2l1HTzP4Ou5apeq2vqsSn92v/4azp1/tRoBwsAAAAAAABfKHIAALaM3/gnv6Fj/80xvfsH7+rjn3yse//hniRp94HdevI3ntQz/+AZ7Xhix4hHCQAAAAAAAL8ocgAAtpSdizv1m//Lb456GAAAAAAAAIgAe3IAAAAAAAAAAIBEosgBAAAAAAAAAAASiSIHAAAAAAAAAABIJIocAAAAAAAAAAAgkShyAAAAAAAAAACARKLIAQAAAAAAAAAAEokiBwAAAAAAAAAASCSKHAAAAAAAAAAAIJEocgAAAAAAAAAAgESiyAEAAAAAAAAAABKJIgcAAAAAAAAAAEik1KgHAABAEhiGsfHe87zA523+vNM1+l3fb/8AAAAAAABbCTM5AADoo1VgaBUX2gsWfs/zPG/jFaSdYRhd2wIAAAAAAGxlzOQAAMCHVnHB87yuRY4g5/lt1ypwBLF6fVUr11ckSYVjBeWX8h3PW3w90GU33Hqlf9tbr3Q/1q1trzb92vdrO2iffozie3VqN+z+4uwzyvu2+VpBn5ewz9cg7eO4F1E/o2HH6Ocag8YUAAAAwNZBkQMAgCEJu+SU33aXly/r5pWbSm9LyzANeY3muU7F0aHnD2n5reUQow5n8fXOScheicvWsTBJz279DdqnX4P20a192O/VS9g49mobtl2vtlHeN7+xChvTQUQd02H/7QEAAADAqFHkAABgCDotUeW30NG+t0d7u9vXbuvCyQvK7slq4cSCMrMZGaahhtOQXbJlrVv66J2P9Nr0a3r5Zy+rcLwgqX9C008CuVcytVeSNmy7Tm3D9hekTz/C9tFthkHQeARNRAeN46Dj7NRn+3X9nB+0zzCiimk/YWMatt2g8Qwbl159x9UOAAAAwNbBnhwAACTY7Wu3dem3Lym/lNf+7+zXvmf2qXCsoNyRnPZ+c692H9ituf1zmj84r+zjWV187qI+efeTWMfUL0katBAh9S68hEms+23rVxR9tJ/n53uFTaqHiWO38/wuxRT0vkd134IW7UaRVA8a0zDtBonnqOICAAAAAH5Q5AAAwIfWklH99tnwe15U7S6cvKDdX9ut/FJehWMFPfHsE8o/ndfeb+3Vrqd2aW5xTjv27dBsblbb89uVfiytN068EaiPJEnasjp+CwDD/l7t/Y3rOP0axwR92Jgm/V4AAAAAQNRYrgoAgD5am4G3ChDdlo/qd177Nf1cv30j8s3HLi9fVnZvVvMH57XnG3uUW8ppbnFOdbuu8qdlpWZSMgxDDbehul2Xa7lyKo6sB5bePvu2Tv/o9MCxAXqJas+TQfsfVX/jWFwZlbCxIaYAAAAA+qHIAQCAD932z2j/3O95QY53O3bzyk0tnFhQ9vGstu/brrnFOe1c2Clr3VLdrsupOLKLtjKzmeYrm1E6m9bMrhm9/9b7sRU5Bt0Qe5hJzGEkwflF/ReiSlj7iemoCyzjMgY/RrnhepRLrgEAAADYmihyAACQQKvXV5XellZmNqP0trTMtKm6XZe1bsm1XHmNh7NDpgyZKVNm2pSZMZXKpJqvmZTWbqwpdzQ30DjCbBrdq22vJXgWX++8MXIUCdphJEzHISkbdxy76XbtQRPWg+zHErVhxjMuccUrbGwmIaYAAAAA4seeHAAA9PD1r39dq6urG6+WzZ+F/fwv//IvN45fvXo10PufvvlTGaYhwzTkNTw5ZUflT8sqrhRVuVuRXbLlWq4abkNew5O8h0tmmZIx1Wyz8t5KRFGKjt9f6G9+DdpXnInwcf7VeVRxDKK1gfUgG1n7iemwk+H9NnIf5+R83M9o2NgkOaYAAAAAhouZHAAAdPGDH/xAKysrHZeLCrosVb9rBH3f/EBqOA05JUfVz6pKzaTkVBwZhiHXclV7UJNTduRarupOvVnwqHvNokdEuiVGu80W6Ne2V7tuGyqH2Wh5Kxc4ooxjmH7bP/PzrLQEKXCMQ9xb329cjTJWYWMz7jEFAAAAMHwUOQAA6KFQKMT2+bPPPrvx/tSpU8Hev3BKF/7wguySrep6VdN3pmUYhuyiLWOqudm4U3ZUe1CTXbblVB4tdhimocLxzmONQtDkdZB2gyZkt3KBY7NxHlsnQWPaKxGehPsTN2IAAAAAYFJQ5AAAIIHyS3k5FUfWuqXK3YpS0yk13IYysxmZKVNew5NrubLLtmr3a7JLDwsdFVeu1Xzljgy2H8c48ZuwpcDR27iOfVzHlVTEEwAAAMAkYU8OAAAS6tDzh5p7cNypqLRWUnGl2HytFlVaK6l8p6zK3Yqqn1dVu1/bmNVh3bd0+OzhUQ9/6MaxwNFviahxSUaHHWcU4w4T026v9nPi1ms2yaAxDfvMjMszFXbJKZaqAgAAANCOIgcAAAm1/NaynIqzUdQorhRV/KRZ6CitlVT+ZVmVOxVV71VV/axZ6LDuW1JDOn3pdKxj65eADdrOzzVHvU/DoH20f7+4krmDFlTCjrPTeUlJyPfi51kfVkz9/n0NK55hYxNFTAEAAABsHSxXBQBAgp37+Tn98Lkfqni7KKfiKJ1NK5VJSabk1T3Vnbrcirsxg8PzPJ27di6y/vslVYMkMPu189u2Vxs/v6wPY5A+Nm+k3Klt0Fh021DcT9tewo6zX7tuhnHfevXZ6/P2PsMWpOKKab97HjSew37WBmkHAAAAYOthJgcAAAmWP5bXi3/yosp3y1r/eH1jyarSamljNkf5TlnF1aKckqOX3nlJuaX49+LothxQv4R0r2WEen2e9F91x7XJetC+/NyfIJ9H0ee46zX+UcV0XISNzaAxBQAAALC1GJ7neXF2cP78eXmep1dffTXObkZikr8bkolnMpm4b4jKle9d0Qc//kCpmZS8RvM/74ZpyLVcHT57OPYlqgAAAAAAAIaN5aoAAJgQZ948ozNvntHajTWtvLciSSocLyh3JP6ZGwAAAAAAAKNAkQMAgAmTO5pT7iiFDQAAAAAAMPkocgAAALQJsukx+wMAAAAAADA6bDwOAAAAAAAAAAASiZkcAAAAbZidAQAAAABAMjCTAwAAAAAAAAAAJBJFDgAAAAAAAAAAkEgUOQAAAAAAAAAAQCJR5AAAAAAAAAAAAIlEkQMAAAAAAAAAACQSRQ4AAAAAAAAAAJBIFDkAAAAAAAAAAEAiUeQAAAAAAAAAAACJRJEDAAAAAAAAAAAkEkUOAAAAAAAAAACQSBQ5AAAAAAAAAABAIlHkAAAAAAAAAAAAiUSRAwAAAAAAAAAAJBJFDgAAAAAAAAAAkEgUOQAAAAAAAAAAQCJR5AAAAAAAAAAAAIlEkQMAAAAAAAAAACQSRQ4AAAAAAAAAAJBIqVEPAAAAJJNhGBvvPc8LdV63Y5s/bz/e6xgAAAAAANhaKHIAAIDAWoUGz/NkGIYMw+hYaOh1Xnubzf/2WwxpPwYAAAAAALYWihwAACCUzQWJXoWGbuf5nX3RrYDS79hmq9dXtXJ9RZJUOFZQfinf8bzF130N6UtuvdK/7a1XOn/ut89u7btdo9f5QXUbo98+wo6vvd2w+4urz0Hj2et6/a4RNKZhn+te7eO4F1E/o2H68tN3FH/vAAAAAB5FkQMAAIxMmKJHEJeXL+vmlZtKb0tLhqSHXTgVR4eeP6Tlt5Yj77ObxdejT1z2Spi2jg3a56B9dGvfKx5hi039xhr1OMP0GfU98xursDEdRNQx7dZukJiOIi4AAAAAokWRAwAAjEy35ap6febn2O1rt3Xh5AVl92S1cGJBmdmMDNNQw2nILtmy1i199M5Hem36Nb38s5dVOF6Q1D8R6if53CsJ2ylJ6yfZ2y+xG7TPoPrFxU/yefNxv2NrHQuaiO5WWIhrnJ36bL9uv3OD9hdWVDHtJ2xMw7YbNKZB4hL2b3bQtgAAAAA6M0c9AAAAgCjdvnZbl757SfmlvPZ/Z7/2PbNPhWMF5Y7ktPebe7X7wG7N7Z/T/MF5ZR/P6uJzF/XJu5/EOqY4k5ZhkqlRXD9IH0EKO63jYZPqQRLg/c7zuxRTkHsQRTz99t9+7VEkz4PGNEy7QWI6qrgAAAAAiA5FDgAAEEprqal+G393Oy+uDcMvnLyg3U/tVn4pr8Kxgp549gnln85r77f2atdTuzS3OKcd+3ZoNjer7fntSj+W1hsn3ohlLINI+jI6fgsAw/6e7f2N6zj9GscEfdiYJv1eDDKucf1OAAAAQBKwXBUAAAistYl4q1DRbdmpXuf124g8zFJVl5cvK7s3q/mD89rzjT3KLeU0tzinul1X+dOyUjMpGYahhttQ3a7LtVw5FUfWA0tvn31bp390OmRE4jOOSWyMfmmhUReHeC67GyQ2xBUAAAAIjpkcAAAgFM/zNl7tn/s5z8+xXn13cvPKTe16apeyj2e1fd92zS3OaefCTm3bs00zczOa3jGtzGzmi1c2o3Q2rZldM3r/rff9fO1Q4khI9/tV+zCS8Pz6/AuLrz/6CnuNIOeNMiE+yPccpiSMEQAAAMBgmMkBAAAmwur1VaW3pZWZzSi9LS0zbapu12WtW3ItV17j4aySKUNmypSZNmVmTKUyqeZrJqW1G2vKHc0NNI5eSVW/SWm/Sexbr/RONg8rCT4Ovz7fHItuG4/HIY4iU9gN7uMyzHjGJe54DXK/x6FoBQAAACQZMzkAAEAgq6urj7zCfH7jxo2N41evXo3k/U/f/KlkSIZpyGt4csqOyp+WVVwpqnK3Irtky7VcNdyGvIYneQ/3BTElY6rZZuW9lQGjs3WMc2I2ilkVQbU2sA67kbXfeA67wNBv8/RxLniM8zMKAAAAIDrM5AAAAL6dP39eBw4c0KlTp750rNsSUv2WoorqffMDqeE05JQcVT+rKjWTklNxZBiGXMtV7UFNTtmRa7mqO/VmwaPuNYseEemWUO02y2AQvZK4cfTnp99R6raEV9wbVncrBPi9B0ELHOMQ99b3G1fjFCsAAAAA8aLIAQAAAvnwww/1wgsvfOnzQqHQ8fxOny8tLW2831wwGej9C6f0iz/8heySrep6VdN3pmUYhuyiLWOqudm4U3ZUe1CTXbblVB4tdhimocLxzt8hCn6T3lElvIMk2YNIQvJ4nMfWLkw8exUXknB/4jbsGLBUFQAAADBaFDkAAMBEyC/l5VQcWeuWKncrSk2n1HAbysxmZKZMeQ1PruXKLtuq3a/JLj0sdFRcuVbzlTsy2H4cky7JCdlxHPs4jinpiCkAAACw9bAnBwAAmBiHnj/U3IPjTkWltZKKK8Xma7Wo0lpJ5TtlVe5WVP28qtr92sasDuu+pcNnD496+BO1/E+/JaLGJRkddpyDjjvM92/f96PTHiBh9wQJqteFtfO9AAAgAElEQVSzOmhMwz4zo3imBvmbHee/dwAAACBJKHIAAICJsfzWspyKs1HUKK4UVfykWegorZVU/mVZlTsVVe9VVf2sWeiw7ltSQzp96XSsYwuS0Ixi6aIoE6iDJo/bxxJXcnfQgkrYcXY6r99+KX7GM2p+nq1hxbRfu1HHdJB+x/05AAAAAMYdy1UBAICJ8vLPXtbF5y6qeLsop+IonU0rlUlJpuTVPdWdutyKuzGDw/M8nbt2LrL++yVjo0pobt742c+v6sPafO2g/fQbY79f5Pf7vFcBIYiw4/R7D7qNL8771qvfXp+39xu2IBVXTPvd86AxHeRZAwAAADAemMkBAAAmSuF4QS/+6Ysq3S1p/eP1jSWrSquljdkc5TtlFVeLckqOXnrnJeWW4t+Lo98yQoMsXxSmv2HpNb5h9tWvv7DjHPf4D6LX+EcV03HChuMAAADAeGAmBwAAmDj7ntmn79e+ryvfu6IPfvyBUjMpeQ1PkmSYhlzL1eGzh30vUeUnETlosnJcl7uJ4tpBrzGqWIRtG7QwFYewhYNBrxtX+2HFNIltAQAAADyKIgcAAJhYZ948ozNvntHajTWtvLciqTnTI3ck/pkbAAAAAAAgfhQ5AADAxMsdzSl3lMIGAAAAAACThiIHAAAxMgxj473neYHP69feT7t+fWNrCbKRNEvqAAAAAADGHRuPAwAQk1ahoVVgaC88+DnP87yuBQrDMDaOe57XsbDRqz0AAAAAAEDSMZMDAIAYtQoMnYoQYc7r1CYqq9dXtXL94b4VxwrKL+UjvT7GA7MzAAAAAACThCIHAAATyu9SWZeXL+vmlZtKb0tLhqSHpzoVR4eeP6Tlt5ZjHikAAAAAAEA4FDkAAJgAraWrWtqLGu3HJen2tdu6cPKCsnuyWjixoMxsRoZpqOE0ZJdsWeuWPnrnI702/Zpe/tnLKhwvDOW7AAAAAAAA+EWRAwCAhOtUwOjn9rXbuvTdS8ov5TV/cF7Zx7NKb0vLa3hySo6q61VV7laUmc2ouFrUxecu6sU/fTGmbwAAAAAAABAORQ4AAGLUKkD022fD73nd2gV14eQF5Zfyyi/ltecbe7R933aZaVNO2VH1s6qm70wrNZ2SOWX+/+zdbYxcV37f+d+99dAtVjfZZJNiVbXUHM1I1IxprqkeUp7JDE2uZxwhi2iyJilYIwGWQSH2ButdYTfxYjeJVhYgB5h9YUPZODZ2ByKcENIMaSpYOEHGYTwawbLsKKLpYAQqloeWOCar+Kzurqdb93FfFKvVbNXDrVvP3d8PUFD1vefcc+6pW5zB/1/nHElS4XJBrxx8RfrHHTcFAAAAAADQNyQ5AADok3rSop64WJ2MWJ2caFdu7fv6+frfjfbeWJssWX3N08dOK7Ujpdnds9r++e1KL6Q1s2tGnu2pdL2k+GRchmHId315tifXcuWUHVnLluzTtsQWHQAAAAAAYESQ5AAAoI+azbJYezxsuV6cu3DmguYPziu1M6XpuWnN7JrRlvktshYtebYnp+zILthKTiVrr1RSiVRCk1snZV+wm14XAAAAAABg0MxhdwAAAAxO/lxeiU0JJaeSSmxKyEyY8mxP1qIl13IV+HdmlcQMmXFTZsKUmTQVT8YVT8aluGRc7WxJLQAAAAAAgH4hyQEAQB/k8/m7XnVvvvnmUN/nzuUkQzJMo7bJeMlR6XpJhVxB5Ztl2UVbruXKd30FfiAFd5a+MlVLfBimlOt8PAAAAAAAAPqB5aoAAOixF198UQ899JAOHz78qXOrl5Ea1nsFku/4coq1Tcbjk3E5ZUeGYci1XFWXq3JKjlzLled4tYSHF9SSHgAAAAAAACOEJAcAAH3wwQcf6KmnnvrU8dWJj2G8z+7PSpLsoq3KYkUTNyZkGIbsgi0jVtts3Ck5qi5XZZdsOeU1yY4gUJAl2QEAAAAAAEYDSQ4AADaQzEJGTtmRtWipfLOs+ERcvusrOZWUGTcV+IFcy5VdslVdqsou3kl0lN3anh1uIKWHfRcAAAAAAAA1JDkAANhg9jyxRxfPXqwlNmK1jceTU0mZCVMKJM/x5JQd2cVaoqM+q8NasqQ9w+49AAAAAADAJ0hyAACwwRw7dUwvTbykQr4gSXItV8lUUmbSlGHUlqxyLVdOubZsVXWpWktw+JKOSWK1KgAAAAAAMCLMYXcAAAAM3vG3jsspOipcLqiQK2g5t6xirqhCvqDi1aJKN0oq3yircrui8u2ygiDQs28/O+xuAwAAAAAA3IUkBwAAG1D2QFZPf+9pFW8WtfjRogq5WrKjmC/WkhzXSirdKKmQL8gpOnrm7DNKL7AZBwAAAAAAGC0sVwUAwAY19+icnq8+rzPfPKP3vvue4pNxBX5tLSrDNORarvY+uVdHXj0y5J4CAAAAAAA0RpIDAIAN7uhrR3X0taO6ev6qcu/mJNVmeqT3MXMDAAAAAACMNpIcAABAkpR+JK30IyQ2AAAAAADA+CDJAQAAIjEMY+V9EASRykU9BwAAAAAAILHxOAAAiKCegKgnH1YnJMKWMwxDQRCsvMKeAwAAAAAAqGMmBwAAiKSeuGiXhGhWrtXsjCgzN/Ln8sqdu7OnyP6sMguZhuV2vdzxpSVJl55rX/fSc42Ph22zWf1W1wtbp5NrrhalT53UXVtv0O31q81ux7PV9dpdo9Mxjfpct6rfj8+i189olLbCtt3rzx8AAABAcyQ5AADA0PRiuarTx07rwpkLSmxKSIakO0WdsqM9T+zRsVPHetrnVna93P8gZtQkTdRr1s9FCQi3Go+o99Gur73uZ5Q2ux3PTtqPUq6Xej2mzep1M6aDHpdef/4AAAAAWiPJAQAAhmZ18qK+RFWYc5J0+e3LOnHohFLbU5o/OK/kVFKGach3fNlFW9aipYtnL+qliZd0/K3jyh7ISmofCA0TfGwVhG0UpA0T7B120LPduIQJPq8+367e2jqdBqKbJRb61c9Gba69bruynbYXVa/GtJ2oYxq1Xrdj2sm49OI72+m/EwAAAACiYU8OAAAwdi6/fVmvPv6qMgsZPfD1BzT36Jyy+7NK70trxxd2aNtD2zTzwIxmd88qtTOlk4+d1JV3rvS1T4MIWPYrIRImMNzpNcIEgKMG1TsJgLcrF3YppmblOu1LL++50bWHETjvdEyj1OtmTAc9Lq3aI7EBAAAA9B5JDgAAEEl9Oal2m4I3K9eqXrtrnjh0Qtse3KbMQkbZ/Vnd96X7lPliRjt+Yoe2PrhVM7tmtHlus6bSU5rOTCtxT0KvHHwlzG0NVJRldEYpSBo2ATDs5YJGtZ9hjdJnXhd1TMf9sxjVfgEAAAAbGctVARg7Ydfpb1Yu7B4Aa8u0OgdsNPVNxOvfi2ZLS7Uq12oj8lbnTh87rdSOlGZ3z2r757crvZDWzK4Zeban0vWS4pNxGYYh3/Xl2Z5cy5VTdmQtW3r9ydd15DtHejwa3Yu6STP6a9hLiQ07OTSKyZVRwdgAAAAAo4MkB4CxsjpQWg+cNko0tCrXSdKi04QIsJE0S/KtPd4qGRjl3IUzFzR/cF6pnSlNz01rZteMtsxvkbVoybM9OWVHdsFWcipZe6WSSqQSmtw6qR+e+mHfkhz9DEgPM9hOcuUTvUgCdLqJ+DCD6aPQhzDG6RkdlzEFAAAAxglJDgBjZ+0vxLst10yzBEq7c3WFKwVdeeeKLv3xJUnSrp/ZpbkDc5qem+64LwBq8ufySmxKKDmVVGJTQmbClGd7shYtuZarwL+T2IwZMuOmzIQpM2kqnozXXpNxXT1/VelH0l31o1VQNWzwstNg57CDosNuv96HXS833rR5EEmmZsejjE3UDe77ZZDj2S/9Hq8on3ejcRyF7xIAAACwnpDkAIAeq3xc0Wv/3WvK/5e8DMNQLBFToEDv/u67UiBlHsno6e89rYnNE8PuKhDJww8/rHw+v/J3JpORpLuONTv+0Ucf6ctf/rIk6c0339ShQ4c6ej/1l1OSIRmmocAP5JQcla6X5NmeAj+QXbTlWq5811fgB1JwZ9aVKRmxWp3cu7mukxyDNMxg8yj/6nwY49JtIiDseA763pptnl7vR6Ok0qgY5We0mVEeTwAAAGAckeQAgAaizuL4r6//V51+8rS23L9F93/5fiWnkzJMQ17Vk120ZS1auvVXt/St2W/pF878gh7+xsP9vA2g51544QXlcrmG34Gwy1c1Ot7R+0DyHV9O0VHldkXxybicsiPDMORarqrLVTklR67lynO8WsLDC2pJjx5pFqBsNssgqlFYpmrUgrHNNqbu94bVrRIBYT7zThMcozDuqxMdo2iUxqqZRn3r9b8TAAAAwEZHkgMAeuTC6Qv6t7/yb3X/l+/X1s9t1VR6SolNCQVe7dfllY8rKt8oK5FKqHStpH/zi/9Gf+/E3xt2t4GOZbPZyMdXvz98+HDH7/Oba7NC7KKtymJFEzcmZBiG7IItI1bbbNwpOaouV2WXbDnlu5Mdhmkoe6BxP3shbNC70+Bsq0BzPwK94xo8HlW9Wuaom+utN4Meg16210lyDAAAAEB7JDkAjJ36TIp2+2yELdcLruXq95/8fd3/lfu186d2ascXdmg6Oy0zYdaCsbcqSl5PKpaMyYgZtV+iu75OHT0lPS+JPcyBUDILGTllR9aipfLNsuITcfmur+RUUmbcVOAHci1XdslWdakqu3gn0VF25Vq1V3rf+CxVNQzjHEAfxb6PYp/GHWMKAAAAYDWSHADGSj1pUU9crF7KZvUyUu3KrX3f7DprNTv33f/+u9p832Zt/exWbX94u9L70pp5YEZuxVXpekmxZG1fDt/15VU9uRW3Fqj92FL1tar0VNQRATaePU/s0cWzF2uJjVht4/HkVFJmwpQCyXM8OWVHdrGW6KjP6rCWLO19cu+wu9/R8j+9nA0SRpTN0Fv9In1UgtFR+9ntck1R7n/Qn3krre692zGN+swM45ka5SW7AAAAAEjmsDsAAJ0KgmDltfZ42HJrX62u0+6c7/j60dkfaeYzM0rdm9JUekqb79usqfSUJrdOKjmdVHIqqWQqqUQqUXttqr0mt05KfyWpd1sFAOvesVPH5JQdFfIFFa8WVcgVVLhSUCFX+7t0raTyjbIqtyqq3K6oulSVtWRJvnTk1SN97VuvEhjD0G3weO299ysw3Oy6UTf1DtvPRuVatTkqCZ522o2nNLgxbVdv2GPai+XlwpwHAAAA0BlmcgBAl3LnckpMJpScSipxT0JGzJBTdlS+VZZX9RR4gWRIhmnIjJky46ZiiZhiydpLCcnIsV4V0Injbx3XycdOqnC5IKfsKJFKKJ6MS6YUeIE8x5NbdldmcARBoGfffrZn7bcLUo56YHut1ffT7tf7jY7V6zSq2+4X+e2Ot0ogdCJqP9vVa6Sb8exG1DGNGnTv15i2+8w7HdNunrWowozpuP07AQAAAIwqZnIAGAuFQkE/+tGPdOnSpZVXXf3vs2fPrhz7gz/4g4G9P/uvzso0Tcmo7bNhF22VbpRUzBVVvllWtVCVW3HlO758z6/N2jC0kviIGTEpH2FQgA0seyCrp7/3tIo3i1r8aLE2myNXUDFfXJnNUbpRUiFfkFN09MzZZ5Re6P9eHJeeG63lhgalVTB7kG21ay9qP7tpc9S16v+wxnSU9HrJsfr5UbtPAAAAYJwxkwPAWPjN3/xN3X///frGN77xqXOpVEqSlE5/EsCcm5sb2PttO7cpH+TlVb3aJuO3K4pPxGWXbBmGIa/qrewJ4FquPNuT59RmeAR+oECBlAg/FgBq5h6d0/PV53Xmm2f03nffU3wyrsCvrf1mmIZcy9XeJ/eGXqIqTNCx28BkLwObo3atTq/RTZvDqNvLIHdUURMH3V63X/UHNaaj/rwAAAAA6A5JDgBj48c//rG2b9/+qeP1Y6vPLSwsDOz9l498We99671aguPjipLXk5KkZCEpM27Kd3w5ZaeW6CjacsrOXckOP/ClbMhBAPApR187qqOvHdXV81eVezcnqTbTI72v/zM3AAAAAADAcJHkAIAu3bv3Xnm2J+tjS+WbZcUn4vJdX8mppGKJmAI/kGvV9gaoLlVVLVTllBw5ZUdexZM8KdjBzuNAt9KPpJV+hMQGAAAAAAAbCUkOAOiBfb+0TxdOXVByKikzZsqrekpMJRRLxCRJnu3JqTiyi58kOuySrWqhKi20uTgAdKiTjaRZVgcAAAAAMM5IcgBAD3zj29/Q+7//vpavLCtQILfifpLkMKTADeRWXTklR9ViVdXFqqyPLcXvicv5u05tM3IAAAAAAAAAHSHJAQA98it/8Sv69qPf1vKPl+WWXSVSCcUmYjIMQ4EfyLM9uZXaslWVj2ubk//yu7+s33rlt4bddQDrDLMzAAAAAAAbhTnsDgDAejHzmRn98ru/rMAItPTjJS3/zbIKuYIK+YKKV4sqXSvV/nu9pMQ9Cf3Kn/+KNt+/edjdBgAAAAAAAMYWSQ4A6KHN85v1a9d+TY/+j4/KrbpavrKs0rWSStdLKl4ryvd8/fT//NP6h/l/qOm56WF3FwAAAAAAABhrLFcFAH3wtW99TV/71tdUvlFW7t2cJCl7IKtN2zcNuWfDYxjGyvsgaL4JSatyUc6tPt7uXLu+AQAAAAAAYLSQ5ACAPtq0Y5Me/DsPDrsbQ1dPJgRBIMMwavuUNEgmtCrX7tza5MXqv5slLlolSgAAAAAAADD6WK4KADAQ9YRCu5kSrcqFvUYUzRIvjdz64JZufXCr530AAAAAAABAZ5jJAQBY98IuldXK+f/3vP70t/5Ut350S/Fk7X8+XdvV7IOz+lv/6G9p3/F9PekrAAAAAAAAwiPJAQAYe/UlrJqdW63RjI1Wszjcqqtv//S3tfQ3S7pn6z2a2z8nM2nKd3zZRVulGyX94f/6h3rnt9/R33/n78uIseQVAAAAAADAoJDkAACsC+02FI+iWqjqtx/+bcWSMe366i5NpaeUSCUU+IHciitr0VJqR0qlmyV9/OHH+q3539KvfvCrPWkbAAAAAAAA7ZHkAAAMRH22RLsERKtyYa7Ryd4a7fzOT/6OEqmE7vvSfZp9eFbT2WnFkjG5Vi3BUb5ZVmJTQmbSlGEYWvqbJf3u3t+VnulJ8wAAAAAAAGiDJAcAoO/qiYl6cmLtrIvVG4o3K9fuGqvLrbY2IRJ2qarv/9Pvq7pc1We//lnt2LND6Z9Ka/P9mxX4gSq3KipeL8qMm1Ig+a4vz/bkVl0tXVqSfiDpUNjRAQAAAAAAQFQkOQAAA9FsdsXa461mYYS9Rthzzc77rq93f/ddze6e1fTctGZ2zWj24Vlt/exWVW5XZJhGLalRduWUHNkFW9VCVcmppCa2TMh5x5HxM+zNAQAAAAAA0G/msDsAAMCo+fivP5ZbdjW5dVIT0xNKbErIMAw5ZUeBH8gwDZkJU2bCVCwZU2wipvhEXPGJuBL3JCRHCj7uzZJZAAAAAAAAaI4kBwCgrwqFwl2vW7durZy7dOnSSL7Pn8tLhhRLxCRJbtVV+VZZhVxBlVsVOSVHnu0p8IPaTBBDMkxDRsyQGTdlGqaUazs0AAAAAAAA6BJJDgBA3/ze7/2ezp8/r3w+v/J6//33V86/8cYbI/m+fLMswzTku76ciiPrY0vFq0UVcgWVrpdU+bgiu2DLrbjybE++48v3fMmvLX9lypRRZrkqAAAAAACAfmNPDgBA33z00Uf68MMP9eu//usrx3bv3r3y/pd+6ZdG8v3O/2anAj+QXbJVXayqdKMkwzRkF22ZMVOe7dXOLVfllBy5Vi3Z4TmeAjeQF3gKdrJcFQAAAAAAQL+R5AAAYI3MFzPybE/VparKt8qK3xNX4AdKFpIy46YCL5BruaoWqqouV2UXbTnlWrLDrboKvEDGHDM5AAAAAAAA+o0kBwAAaySnkrr/y/fr1l/d0sT0hMy4Kc/xlEwlZSZMKZA825NTdmqJjqWq7IItu2TLLtrSvBTEmckBAAAAAADQbyQ5AABo4OdP/rz+xe5/oeL1omTUNh9PppKKJWubkfuuL9dyV5atqic6XMuVjg658wAAAAAAABsEG48DANDAlvkt+pkXfkbFfFGFfEGF3KpXvqDi1aJK10sq3yyrcruiymJF1UJVP/sbPytND7v3g2EYxsorarmo51aXAQAAAAAAGxczOQAAaOLg/3FQicmEvv9Pvi+7YGtiekKxiZgM01AQBPKdO7M5CrYcy9Hf/r/+tg786gGdffHssLved/XkQhAEK4mIIPj0El2tyq2ts/Zcu+uT4AAAAAAAACQ5AABo4Uv/y5c0/5V5nf6F0ypeLcpzPcUStSWrPLv2PrU9pV/8j7+o9EJ6yL0drHrSoZ6I6LRco6RI2OvXkx5hEx35c3nlzuUkSdn9WWUWMg3L7Xo51OU+5dJz7eteeq7x8bBtNqoftc1ONWsn7PUb1Q9Td229QbfXrza7Hc9W12t3jU7HtNtnbFCfRa+f0ShtRW177TV79b0FAAAANgqSHAAAtJF9NKvn/vo5/fhPfqwP/+hD/c3bfyNJuv8r9+uBn31A81+dH3IPx1cnSY/VdcKWPX3stC6cuaDEpoRkSLpTzSk72vPEHh07dazTLke26+XxDF62CujWz0UJCLcaj6jJpnZ97XU/o7TZ7Xh20n6Ucr3U6zFtVq+bMR3GuIxiHwAAAIBxRpIDAIAwDGn+q/MkNHqs2XJVzYRNcFx++7JOHDqh1PaU5g/OKzmVlGEa8h1fdtGWtWjp4tmLemniJR1/67iyB7KS2gdCwwSfWwVhGwVpwwR727Xb7+RJu3EJE3xefb5dvbV1Og0CN0ss9Kufjdpce912ZTttL6pejWk7Ucc0ar1ux7STcenFdxYAAABA77DxOAAAGCtrNyRfu2TV5bcv69XHX1VmIaMHvv6A5h6dU3Z/Vul9ae34wg5te2ibZh6Y0ezuWaV2pnTysZO68s6VvvZ5nAOeYQLDnV4jTMIgalC9kwB4u3Jhl2JqVq7TvvTynhtdexjPYadjGqVeN2M6rHGpIykCAAAAdI8kBwAAiKRZkiFsuSj1giC461U/ttqJQye07cFtyixklN2f1X1fuk+ZL2a04yd2aOuDWzWza0ab5zZrKj2l6cy0Evck9MrBV0Lc8WCN+xI2YRMAg77Pte2Naj/DGsXgeNQxHffPops9fQAAAABEx3JVAACgY/VNv+sJiGbLTrUq12oj8lb1Wjl97LRSO1Ka3T2r7Z/frvRCWjO7ZuTZnkrXS4pPxmUYhnzXl2d7ci1XTtmRtWzp9Sdf15HvHIk4Iv3Tzw26Ed2wf4E/7OQQz1hz3WxWDwAAAKBzJDkAAEAkzRIPa4+3SlBEPdeszIUzFzR/cF6pnSlNz01rZteMtsxvkbVoybM9OWVHdsFWcipZe6WSSqQSmtw6qR+e+mHfkhzDCkj3OxBNkPYTvUgCdLqJOMsstTeqz+i4jB8AAAAwDkhyAACAdSF/Lq/EpoSSU0klNiVkJkx5tidr0ZJruQr8O7NDYobMuCkzYcpMmoon47XXZFxXz19V+pF0V/1oFVTt9BfeUTY6H0ZQdxQCtZeeq917ow2m+zkmza7dTRA76gb3/TIKz1i3BpXw66SdUfjeAAAAAOsBe3IAAICO5PP5u15hjl+7dk1vvvnmyrF+vP/Baz+QDMkwDQV+IKfkqHS9pEKuoPLNsuyiLddy5bu+Aj+Qgjv7fZiSEavVyb2b62JkBq/dxtaDCO6PYqC2nuyovwahvoF11I2sw47noBMMw3zGujWqz+gojxkAAAAwjkhyAACA0F588UW98cYbn9r8W2q8KXj9+Or/9ut97YDkO76coqPK7YqKV4sq5AoqXSupcqui6nJVTsmRa7nyHK+W8PCCWtKjR9YGu1cHvQcZcO+nUQ0eN0swRE08dNJus2NhPvNOExyjMO6j0IdWRmmsVhvVfgEAAADjjOWqAABARz744AM99dRTnzqezWYblq8f37lz58qxw4cP9/79U4f14e98KLtoq7JY0cSNCRmGIbtgy4jVNht3So6qy1XZJVtO+e5kh2Eayh5ofA+90Go5pdVGPQg66v2TRrtva0UZz1aJk3H4fPpt0GPAZwgAAAAMF0kOAACwLmQWMnLKjqxFS+WbZcUn4vJdX8mppMy4qcAP5Fqu7JKt6lJVdvFOoqPsyrVqr/S+7vbjWO/GOfg6in0fxT6NO8YUAAAA2HhIcgAAgHVjzxN7dPHsxVpiI1bbeDw5lZSZMKVA8hxPTtmRXawlOuqzOqwlS3uf3Dvs7vdsOat+LIvVafC43cyVUQlGR+1nvV5Uvd6oelizFxrpdkyjPjPDeKY6fQZG6TMEAAAA1gv25AAAAOvGsVPH5JQdFfKFlf04ClcKKuRqf5eulVS+UVblVkWV2xVVl6qylizJl468eqSvfeskGBomyNnsequP9ypY2m3wdW1f+7U3Sbsx6XRT77D9bFSuVZvjEszuxTPWqzFtV2/YYzrqnyUAAACwnjGTAwAArCvH3zquk4+dVOFyQU7ZUSKVUDwZl0wp8AJ5jie37K7M4AiCQM++/WzP2m8XjO1lMHQQG5mvbqPdr/cbHavXaVS33S/y2x1vlUDoRNR+tqvXSDfj2Y2oYxr1GevXmLb7zDsd026eNQAAAACjgZkcAABgXckeyOrp7z2t4s2iFj9arM3myBVUzBdXZnOUbpRUyBfkFB09c/YZpRf6vxfHped6u1RNq3Lt2hqkVsHsQbbVrr2o/eymzVHX7TPWjzEdJcOePQIAAACghpkcAABg3Zl7dE7PV5/XmW+e0XvffU/xybgCP5AkGaYh13K198m9oZeoChPE7DbQGaX+IIKrvWij02t00+Yw6vZqX41uRE0cdHvdftUf1JgOq+4grgcAAABsFCQ5AADAunX0taM6+tpRXT1/Vbl3c5JqMz3S+/o/c0xnJo4AACAASURBVAMAAAAAAPQfSQ4AALDupR9JK/0IiQ0AAAAAANYbkhwAAAAbSCcbSbN8DgAAAABg1LHxOAAAAAAAAAAAGEvM5AAAANhAmJ0BAAAAAFhPmMkBAAAAAAAAAADGEkkOAAAAAAAAAAAwlkhyAAAAAAAAAACAsUSSAwAAAAAAAAAAjCWSHAAAAAAAAAAAYCyR5AAAAAAAAAAAAGOJJAcAAAAAAAAAABhL8WF3AACA9cwwjJX3QRB0XK5d/Vbnw7YNAAAAAAAwrpjJAQBAn9STDPUEw+qkQ9hyQRA0TVAYhrFyPgiCu+q1OgcAAAAAALBekOQAAKCP6gmKdjMpwpZrVKfTcwAAAAAAAOsFy1UBALDBfXzxY/3n3/7P+vCND3X74m1J0rYHt+mB//YBPfo/PaqZz8wMuYcAAAAAAACNkeQAAGAdqC9P1em5s//bWf35t/9cCqTJmUlt/cxW+Z6v8rWyzv0/53T+lfPa/w/262v/7Gv97D4AAAAAAEAkJDkAABhzURMc//rn/rXy5/JK70traueUkqmkJMmxHFWXq7JuWyrfLOs//d//SdfOX9NT//6pvt0DAAAAAABAFCQ5AADoo3qSod3G32HLNavX6bmTj53U1f9yVbsO79LsQ7OaykwpPhmXZ3uqLlVVvlVWebqs2GRMRtzQpbcu6dW/+6p0oKPuAQAAAAAA9BVJDgAA+qSetKgnLlYnHFYnINqVW/u+fr7+9+oyYc69f+Z9Xf6zy5o/OK97f/Je7dy7U5vv2ywZkrVoqXS9pPhkXIZpKPAD+Y4vz/b00Q8+kjZL2t392AAAAAAAAPQCSQ4AAPqo2UyKtcfDluvFuTf+zze09TNbtWV+i7Z9bpt2/OQOze6eVeVWRYVcQYEfyLM9ORVHTtmRXbJlF21VU1U5f+SQ5AAAAAAAACPDHHYHAADA4BTzRd2+eFub7t2ke7beo4mZiZW9OMy4qdhETPHJ+Kdf98SVSCWkW5JR6mxJLQAAAAAAgH5hJgcAABtI7lxuJZlhxAz5ri9r0ZIMyXd9uZYr3/WloLbUlWEaMmOmzHjtZcQMBbnms0QAAAAAAAAGiZkcAAD02OHDh/X444/r3LlzK6+6F198cajvF/96UWbMrC1JVa1tMl68WlQhV1D5elnVxaqckiPXcuU5ngIvUOAHK0mPmBGTbnc+JuOmvkdKmA3jG5VbfbzVNaKeAwAAAAAANczkAACgxw4dOtT03AsvvDDU9zOfnanN2Ki4qi5XVb5ZlhkzZRdtmTFTnu3JLtb24HDKnyQ7fNeX7/ryfE/a1n4MxtnqDd7rSYpGe5y0K9dqX5TV9Ts9BwAAAAAAPkGSAwCADST7xax8z1d1uarK7YoSmxIK/EDJQlJm3FTgBXIqjuyCXXuVbLllt5bssD0FfqAgu/6Xq6onKOoJjG7LrVVPiDSq0+pcI/lzeeXO5SRJ2f1ZZRYyDcvtejl09+5y6bn2dS891/h42DbX1o9aL4pmbYW9dqP6YequrTfo9vrVZrfj2ep67a7R6ZhGfa5b1e/HZ9HrZzRKW1Ha7uaZAwAAADpBkgMAgA1kKjOl2QdnVblZUWlLSWbclOd4Sk4lFUvEastY2Z7ski17+c7rzqwOp+JIs5JSw76L8bA6SbF6VkezmSHtzq11+thpXThzQYlNCcmQdKeaU3a054k9OnbqWOS+d2rXy+MZvGwV0K2fixIQbjUeUZNN7fra635GabPb8eyk/SjleqnXY9qsXjdjOoxxadfuuP5bAQAAgNFGkgMAgA3mZ//Zz+rMU2eUmE7IMAy5VXclySFJvuPLsRzZRVvV5aqsZUt20ZZbcaWfH3Lnx8TaRMXq2RndJjguv31ZJw6dUGp7SvMH55WcSsowDfmOL7toy1q0dPHsRb008ZKOv3Vc2QNZSe0DoWECj62CsI2Cl2GCvY3KRK0XRbtxCRN8Xn2+Xb21dToNRDdLLPSrn43aXHvddmU7bS+qXo1pO1HHNGq9bse0k3Hp5Xev06QNAAAAEBUbjwMAsME8/I2H9ZlDn9HSpSUV8gUVcgUVrtz5b76g4rWiyjfKqtyqyPrYUnWxqvKtsj779c9KDw279+Nv7Ybkq2d8tDon1RIcrz7+qjILGT3w9Qc09+icsvuzSu9La8cXdmjbQ9s088CMZnfPKrUzpZOPndSVd6709X7G+VfZYQLDnV4jTMIgalC904RQq3Jhl2JqVq7TvvTynhtdexjPYadjGqVeN2M6jHGJ8twAAAAA3SLJAQDABvTUv3tK81+d19KPl1TMFWsJjlxBxXxRxWtFla6VVLpRUvlmWaWbJX3ua5/Tk//fk8Pu9sA0SzJELVcXBMFdr/qxdufqThw6oW0PblNmIaPs/qzu+9J9ynwxox0/sUNbH9yqmV0z2jy3WVPpKU1nppW4J6FXDr4S/sYHpB9LNg1S2EDuoPu7tr1R7WdYoxgQjzqm4/5ZjGq/AAAAAInlqgAA2LCe/vdP64/+yR/p3X/5rorXiopNxBSfqP1fA9dy5VU9xe6J6Su/9hUdfvHwcDs7QPVlpeqJi2b7abQrt/aa3Tp97LRSO1Ka3T2r7Z/frvRCWjO7ZuTZnkrXS4pPxmUYhnzXl2d7ci1XTtmRtWzp9Sdf15HvHOm6D70WNYg9isHv9aTXS4JFbX9Y7fF8NTfIpeQAAACAsEhyAACwgX3tN76m/b+8X+/8y3f00R99pFt/dUuStG33Nn32a5/Vo7/6qDbft3nIvRy8ZkmJtcfDluu0nUbnLpy5oPmD80rtTGl6blozu2a0ZX6LrEVLnu3JKTuyC7aSU8naK5VUIpXQ5NZJ/fDUD/uW5Fivv/Ber/cVRS+SAJ1uIj7MQPko9CGMUX9GSR4BAABgUEhyAACwwW3ZtUU/962fG3Y30EL+XF6JTQklp5JKbErITJjybE/WoiXXchX4d2aVxAyZcVNmwpSZNBVPxmuvybiunr+q9CPprvrRKqgaNoAZNYA8rMDzKARmLz1Xu/9GG0z3M9Dd7NrdfBZRN7jvl0GOZ7/0e7w6/bz78dwAAAAArZDkAAAAuCOfz9/1dyaT6enx69ev6/3339ehQ4ckSW+++Wao91N/OSUZkmEaCvxATslR6XpJnu0p8APZRVuu5cp3fQV+IAV3lswyJSNWq5N7N9d1kmOjGOVg7DCC8N0mAsKO56Dvrdnm6fV+NEoqjYpRfkbr1kMCCQAAAOOBjccBAAAkvfjii3rjjTc+tfm31HhT8CjH6+civQ8k3/HlFB1VbldUvFrbML50raTKrYqqy1U5Jae2n4rj1RIeXlBLevTIpecav6T1E8Ac1eDx6rEOc7yX7TY7FuYz7zTBMQrjPgp9aGWUxqqZbp8bAAAAoBPM5AAAALjjgw8+0FNPPfWp49lstmH5To/fe++9uvfee1f+Pnz4cKj3+c21mSF20VZlsaKJGxMyDEN2wZYRq2027pQcVZersku2nPLdyQ7DNJQ90LhPvdBqOaXVRn2pqnENHo+qKOPZKgA+Dp9Pvw16DBhzAAAAjAOSHAAAACMus5CRU3ZkLVoq3ywrPhGX7/pKTiVlxk0FfiDXcmWXbFWXqrKLdxIdZVeuVXul97FUVSvjHMwdxb6PYp/GHWMKAAAANMZyVQAAAGNgzxN7VMgVVL5RXlmqqpArqJAvqHi1qNKNkso3y6p8XFF1qboyq8NasrT3yb3D7n7kJWoGsbRNp8HjdsvujEowOmo/u+13lPtvthTa6mv0e2muulbPXLdjGvWZGcYz1el3b9jPOwAAADYukhwAAABj4NipY3LKzkpSo5ArqHCllugoXi2qdK2k8o2yKrcqqtyuJTqsJUvypSOvHulr3zoJhkYNhPYrgNpt8HjtvfcrKdNtQiVqPxuVa9XmqCR42mk3ntLgxrRdvWGPaZR2O31uAAAAgG6wXBUAAMCYOP7WcZ187KQKlwtyyo4SqYTiybhkSoEXyHM8uWV3ZQZHEAR69u1ne9Z+u2DsuAUvV99PmF/vrz1Wr9Oobrtf5Lc73iqB0Imo/WxXr5FuxrMbUcc0akKqX2Pa7jPvdEy7edaiivLcAAAAAN1iJgcAAMCYyB7I6unvPa3izaIWP1pcWbKqmC+uzOYo3SipkC/IKTp65uwzSi/0fy+OdssIjfqG41G1CmYPsq127UXtZzdtjrpW/R/WmI6Sbr576/m5AQAAwGhiJgcAAMAYmXt0Ts9Xn9eZb57Re999T/HJuAI/kCQZpiHXcrX3yb2hl6gKE3TsNjA5aktU9fL6nV6jmzaHUbfTfTX6IWrioNvr9qv+oMZ0WHV7UR8AAADoBEkOAACAMXT0taM6+tpRXT1/Vbl3c5JqMz3S+/o/cwMAAAAAgFFBkgMAAGCMpR9JK/0IiQ0AAAAAwMZEkgMAAERiGMbK+yAIOi4Xpn43dbHxdLLRMcvpAAAAAMD6wMbjAACgY/UkQz3BsDrpELZcEARtkyP1MmsTHKuPN2sbAAAAAACsf8zkAAAAkdQTD+0SDWHLrVZPZLS6Xify5/LKnbuzb8X+rDILmY6vgdHH7AwAAAAA2HhIcgAAgJHUiyWpTh87rQtnLiixKSEZku5cxik72vPEHh07dawHPQUAAAAAAMNCkgMAAIykRktUrdXs+OW3L+vEoRNKbU9p/uC8klNJGaYh3/FlF21Zi5Yunr2olyZe0vG3jit7INvXewEAAAAAAP1BkgMAAIylVgmOVx9/VZmFjGZ3zyq1M6XEpoQCP5BTdFRZrKh8s6zkVFKFfEEnHzupp7/39BDuAAAAAAAAdIskBwAAiKSeZGi3z0bYclHabuTEoRPKLGSUWcho++e3a3puWmbClFNyVLld0cSNCcUn4jJjpiSpcLmgVw6+Iv3jnnUPAAAAAAAMCEkOAADQsXrSop64aLa0VLtya98326S8Ub1G508fO63UjpRmd89q++e3K72Q1syuGXm2p9L1kuKTcRmGId/15dmeXMuVU3ZkLVuyT9sSW3QAAAAAADBWSHIAAIBIms2kWHs8bLmo11/twpkLmj84r9TOlKbnpjWza0Zb5rfIWrTk2Z6csiO7YCs5lay9UkklUglNbp2UfcFu2R8AAAAAADB6zGF3AAAAoBfy5/JKbEooOZVUYlNCZsKUZ3uyFi25lqvAvzOrJGbIjJsyE6bMpKl4Mq54Mi7FJeNq75bUAgAAAAAA/UeSAwAAdOThhx9WPp9fedXl83m9//77K3+/+eabA32fO5eTDMkwjdom4yVHpeslFXIFlW+WZRdtuZYr3/UV+IEU3FnyylQt8WGYUi7SkAAAAAAAgCFhuSoAABDaCy+8oFwu13DJqCAI7jo+lPeB5Du+nGJtk/H4ZFxO2ZFhGHItV9XlqpySI9dy5TleLeHhBbWkBwAAAAAAGDskOQAAQEey2WzT46vPHT58eKDv85trs0rsoq3KYkUTNyZkGIbsgi0jVtts3Ck5qi5XZZdsOeU1yY4gUJAl2QEAAAAAwDghyQEAANaFzEJGTtmRtWipfLOs+ERcvusrOZWUGTcV+IFcy5VdslVdqsou3kl0lN3anh1uIKWHfRcAAAAAAKATJDkAAMC6seeJPbp49mItsRGrbTyenErKTJhSIHmOJ6fsyC7WEh31WR3WkiXtGXbvAQAAAABAp0hyAACAdePYqWN6aeIlFfIFSZJruUqmkjKTpgyjtmSVa7lyyrVlq6pL1VqCw5d0TBKrVQEAAAAAMFbMYXcAAACgl46/dVxO0VHhckGFXEHLuWUVc0UV8gUVrxZVulFS+UZZldsVlW+XFQSBnn372WF3GwAAAAAARECSAwAArCvZA1k9/b2nVbxZ1OJHiyrkasmOYr5YS3JcK6l0o6RCviCn6OiZs88ovcBmHAAAAAAAjCOWqwIAAOvO3KNzer76vM5884ze++57ik/GFfi1tagM05Brudr75F4defXIkHsKAAAAAAC6QZIDAACsW0dfO6qjrx3V1fNXlXs3J6k20yO9j5kbAAAAAACsByQ5AADAupd+JK30IyQ2AAAAAABYb0hyAAAArGEYxsr7IAg6Lrf6eKNrRK0HAAAAAADuRpIDAABglXqiIQgCGYYhwzAaJhvalWuWoFhbLmw9AAAAAADwaSQ5AAAA1qgnGuoJjG7L9VP+XF65c3f2G9mfVWYh07DcrpejXf/Sc+3rXnqu/XUaXSNKvTB1OtHs3sK2M+j76lV7/Wqz2/Fsdb121+h0TLt9rgf1WfT6Ge33s9ZtXQAAAKBTJDkAAAD6oNmSVGsTImGXslrr9LHTunDmghKbEpIh6U5Rp+xozxN7dOzUsS7vILxdL3ce9F59rlHdqEmZTkTtW7v6UcejlXZ97XU/o7TZ7Xh20n6Ucr3U6zFtVq+bMe3Xs9aqzVb1240NAAAAEBVJDgAAgB5rlLioH2u1XFWrenWX376sE4dOKLU9pfmD80pOJWWYhnzHl120ZS1aunj2ol6aeEnH3zqu7IGspPaB0DDBx1ZB2HZB707rrq3Xr0B2u3EJE3xefT7MPa2u0+l9NRvjfvWzUZtrr9uubKftRdWrMW0n6phGrdftmHYyLt1+Z3vxvAEAAACdMofdAQAAAIRz+e3LevXxV5VZyOiBrz+guUfnlN2fVXpfWju+sEPbHtqmmQdmNLt7VqmdKZ187KSuvHOlr32KmhxpV/fSc/0PhoYJDHd6jTAJg6hB9U4C4O3KhV2KqZPPrhfjGbb9tdceRuC80zGNUq+bMe1mXKJ8Z1uVI7EBAACAfiLJAQAAsEZ9yah2+2yELdcrJw6d0LYHtymzkFF2f1b3fek+Zb6Y0Y6f2KGtD27VzK4ZbZ7brKn0lKYz00rck9ArB18ZSN82krAJgEEvo7S2vVHtZ1ijGBiPOqbj/lmEsRHuEQAAAKOJ5aoAAABWqe+ZUU9ctFpaqlW5tddce/1G51rVO33stFI7UprdPavtn9+u9EJaM7tm5NmeStdLik/GZRiGfNeXZ3tyLVdO2ZG1bOn1J1/Xke8ciTwm2Fii7J/Rj/aH1d4oJlcAAAAANMdMDgAAgDWCIFh5rT0etlyzc1HrXThzQVsf3KrUzpSm56Y1s2tGW+a3aNP2TZqcmdTE5gklp5KfvFJJJVIJTW6d1A9P/bCb4WipVUC63S+3hx1Mb4Vfm39i18t3v6Jeo5Nyw3wmurnPQepHH8f5OwsAAICNi5kcAAAAIy5/Lq/EpoSSU0klNiVkJkx5tidr0ZJruQr8O7NKYobMuCkzYcpMmoon47XXZFxXz19V+pF0V/0Ik9BodLxV0HjUg6Wj0L/VY9hqc/de60egu5s9XPphkOPZL70er3H/zgIAAGDjIckBAABwx8MPP6x8Pr/ydyaTkaS7jvXieDKZ1HvvvadDhw5Jkt58882W73PncpIhGaahwA/klByVrpfk2Z4CP5BdtOVarnzXV+AHUnBn6StTMmK1Orl3c10nOTaKUf61+jCC8N0mAsKO56Dvrdnm6fV+NEoqjYpRfkYBAACAQSPJAQAAIOmFF15QLpdrurxUI90c7/S9Asl3fDlFR5XbFcUn43LKjgzDkGu5qi5X5ZQcuZYrz/FqCQ8vqCU9eqRZQLXZLIP6uWZ1W9UbllENHjdbRqjfmzm3SgSE+ew6TXCMwrivTnSMon6P1bh9ZwEAAACSHAAAAHdks9mBHT98+HDo99n9tXp20VZlsaKJGxMyDEN2wZYRq2027pQcVZersku2nPLdyQ7DNJQ90LhPvdAs6N0uGNtJsHwQRinQ3swo922tKOPZKrkwDp9Pvw0zwVE/PkrfWQAAAEAiyQEAADDyMgsZOWVH1qKl8s2y4hNx+a6v5FRSZtxU4AdyLVd2yVZ1qSq7eCfRUXblWrVXeh9LVbUyzgH0Uez7KPZp3DGmAAAAQGPmsDsAAACA9vY8sUeFXEHlG2UVrxZVyBVqr3xBxatFlW6UVL5ZVuXjiqpL1ZVZHdaSpb1P7h1290dap8HjdktEjUowOmo/u+13lPu/9Fzz19oy/dZqNkm3Yxr1mRmVZ6qVcfleAAAAYP0hyQEAADAGjp06JqfsrCQ1CrmCCldqiY7i1aJK10oq3yircquiyu1aosNasiRfOvLqkb72rd3+Be2CnsPUbeB17T306566DRxH7Wejcu32bAjTn2EL80wOakzDfn8GOabdfmcH9b0AAAAAJJarAgAAGBvH3zquk4+dVOFyQU7ZUSKVUDwZl0wp8AJ5jie37K7M4AiCQM++/WzP2m8XqGz06/V6nTC/jg/TVrONt6NYfa1O+9fu3tr9Ir/d8U7GpJWo/Qz72TXrX6fj2Y2oYxo18N6vMW33mQ/iO9TNdzZM/VFPfgEAAGA8MZMDAABgTGQPZPX0955W8WZRix8trixZVcwXV2ZzlG6UVMgX5BQdPXP2GaUX+r8XR6tlhKKeGwet7muQbbVrL2o/1+vnJrXu/7DGdFR0+50dh3sEAADA+sJMDgAAgDEy9+icnq8+rzPfPKP3vvue4pNxBX4gSTJMQ67lau+Te0MvURUm8NiL4GSn1xhEQHTc7msYdTvdV6MfogbVu71uv+oPakyHdX+9aBsAAADoBEkOAACAMXT0taM6+tpRXT1/Vbl3c5JqMz3S+/o/cwMAAAAAgFFBkgMAAGCMpR9JK/0IiQ0AAAAAwMZEkgMAAADrQicbSbOcDgAAAACsD2w8DgAAAAAAAAAAxhIzOQAAALAuMDsDAAAAADYeZnIAAAAAAAAAAICxRJIDAAAAAAAAAACMJZIcAAAAAAAAAABgLJHkAAAAAAAAAAAAY4kkBwAAAAAAAAAAGEskOQAAAAAAAAAAwFgiyQEAAAAAAAAAAMYSSQ4AAAAAAAAAADCWSHIAAAAAAAAAAICxRJIDAAAAAAAAAACMJZIcAAAAAAAAAABgLJHkAAAAAAAAAAAAY4kkBwAAAAAAAAAAGEskOQAAAAAAAAAAwFgiyQEAAAAAAAAAAMYSSQ4AAAAAAAAAADCWSHIAAAAAAAAAAICxRJIDAAAAAAAAAACMpfiwOwAAAEaXYRgr74MgiFyu1flm58K2DQAAAAAANi6SHAAAoKF6kiEIAhmGIcMwWiYwmpVrVq/VuU6uAQAAAAAANi6SHAAAoKl6YqGewOi0XJQER1T5c3nlzuUkSdn9WWUWMg3L7Xo52vUvPde+7qXnGh8P22aj+s3qNmsrqm7baVQ/TN219QbdXr/a7PXntvp67a7R6ZhGfa5b1e/HZ9HrZzRKW2HbHtT3FgAAAABJDgAA0GdRlqpamyxplQw5fey0Lpy5oMSmhGRIulPUKTva88QeHTt1rMs7CG/Xy70NYrYKtNbP9aK9bttpVr/VeERNNrXra6/7GaXNXn9uYccq6ph2o9dj2qxeN2M66HEZ1PcWAAAAQA1JDgAA0Fetlp1qdi7MclWX376sE4dOKLU9pfmD80pOJWWYhnzHl120ZS1aunj2ol6aeEnH3zqu7IGspPaB0DDBx1ZB2EZB2jDB3lZlOm0vinbjEib4vPp82P7Vz3UaiG6WWOhXPxu1ufa67cp22l5UvRrTdqKOadR63Y5pJ+PSj+/s6rr9/PwBAACAjcYcdgcAAAA6dfnty3r18VeVWcjoga8/oLlH55Tdn1V6X1o7vrBD2x7appkHZjS7e1apnSmdfOykrrxzpa996kfA8tJzza/by/aiBGvblQuTMIgaVO8kAN6uXNilmDr5HHoxnmHbX3vtYQTOOx3TKPW6GdNBj0svP38AAAAA7ZHkAAAATdWXjGq1H0cn5XrlxKET2vbgNmUWMsruz+q+L92nzBcz2vETO7T1wa2a2TWjzXObNZWe0nRmWol7Enrl4CsD6VsnhrG8UC+FTQAMe7mgUe1nWKMYGI86puP+WYxqvwAAAICNjOWqAABAQ/V9MeqJi2bLR7Uq12pvjajnTh87rdSOlGZ3z2r757crvZDWzK4Zeban0vWS4pNxGYYh3/Xl2Z5cy5VTdmQtW3r9ydd15DtHuh6bXhvFIDaGv3/CsJNDPJfNMTYAAADA6CDJAQAAmmq24ffa4602Bu/1uQtnLmj+4LxSO1OanpvWzK4ZbZnfImvRkmd7csqO7IKt5FSy9kollUglNLl1Uj889cO+JTmGFZDud7CVX65/ohdJgE43ER9mMH0U+hDGOD2j49RXAAAAYFyQ5ACAEdLsl+udlIt6rpP2gWHJn8srsSmh5FRSiU0JmQlTnu3JWrTkWq4C/86skpghM27KTJgyk6biyXjtNRnX1fNXlX4k3VU/WgUqwwaEOw0gN2pzVPYZGGQfdr3ceNPmfgaPm127myRA1A3u+2WQ49kvg0r49aKdUfg+AQAAAOsFSQ4AGBGrl/qpL/3TKhHRqNzaOmvPtbp+s/awMeXz+bv+zmQyAzm+vLys8+fP69ChQ5KkN9988673U385JRmSYRoK/EBOyVHpekme7SnwA9lFW67lynd9BX4gBXeefVMyYrU6uXdzXSc5RkWjYH+vry+NZkB2GEH4bhMBYcdz0PfWbPP0ej/6/Zx1Y5Sf0bXGqa8AAADAOCHJAQAjZO0eB52Wa5ekaFavkwTH8pVlnfvdc/rr//jXuvnBTUnS9oe363Nf/5z2/4P9mspMhboORteLL76ohx56SIcPH/7UubDLV0U9vvZcw/eB5Du+nKKjyu2K4pNxOWVHhmHItVxVl6tySo5cy5XneLWEhxfUkh490ixI2WyWQb/a7Gd7oxqQbbYxdb83rG6VCAjzGXSa4BiFcV+d6BhFozRW7YxTXwEAAIBxQ5IDANaZqEtOhan3gxd+oHf++TsKgkATWyY0nZlW4AZa/HBRb//m2/qzf/5n+so/+ooO/tOD0W8AI+GDDz7QU0899anj2Wy2YfleHd+8efNdyZW17/ObazNA7KKtymJFEzcmZBiG7IItI1bbbNwpOaouV2WXbDnlu5MdgiIk7AAAIABJREFUhmkoe6Bx270QNujdq4BnJ0H2ToxDQHaU+7ZWlPFslVwYh8+n3wY9Bt20x+cFAAAA9BdJDgBYZ6IuQdWu3muPv6Yf/8mPtfOndmpq55SSU0lJWvnlfOV2ReWbZf3xt/5YuT/P6Rde/4Ue3A1wt8xCRk7ZkbVoqXyzrPhEXL7rKzmVlBk3FfiBXMuVXbJVXarKLt5JdJRduVbtld63Ppaq6pdxDsiOYt9HsU/jbpzGdJz6CgAAAIwrkhwAgLZee/w1Xf7Ty9r1M7s0+9CspjJTik/G5TmeqktVVW5VlJxOKj4Zlxk3dfE/XNSpI6eknxp2z7Ee7Xlijy6evVhLbMRqG48np5IyE6YUSJ7jySk7sou1REd9Voe1ZGnvk3uH3f11tfxPu5kkoxLgjdrPbpdrinL/g5gBFFare+92TKM+M8N4pqI+A6Py/AMAAADrnTnsDgAAPlFfMqrVfhytykWt18oH/+4DXXrzkuZ+ek479+5Udn9W9/30fUrvS2v77u2a+cyMpuemNZWeUmpHSpu2b9J0Zlo/+sMfSRdDNwOEduzUMTllR4V8QcWrRRVyBRWuFFTI1f4uXSupfKOsyq2KKrcrqi5VZS1Zki8defVIX/vWSTA0TOCz3fV6mTDpNiC7ti/9SuY0u27UTb3D9rNRuVZtjkuAu914SoMb07DP+7DGNMpyY6P++QMAAADrATM5AGBE1DcDrycgmi0f1apcq43Io9b7/v/+fW15YIu2zG/R1s9t1b1779XsQ7Oq3K6okC8oCAJ5tifXcmu/ni/ZK7+gd/6DI/0PPRke4C7H3zquk4+dVOFyQU7ZUSKVUDwZl0wp8AJ5jie37K7M4AiCQM++/WzP2m8XjO3H/hj9bG91G+1+vd/oWL1Oo7rtfpHf7nirBEInovazXb1GuhnPbkQd06gJqX6NabvPvNMx7eZZi2JYnz8AAACwUTGTAwBGSBAEK6+1x8OU6/W50rWSbv7VTU3dO6V7tt2jyZlJJaeTMkxDZsJUbCKm+GT87tc9tVdiKiHdlIxy+FkjQFjZA1k9/b2nVbxZ1OJHi7XZHLmCivniymyO0o2SCvmCnKKjZ84+o/RC//fiuPRcb5cbaleuXXuD0iqYPci2woxXJ8d70eaoa9X/YY3pKGFGBgAAADD6mMkBAGgq925OZryWzDBihgIvkPWxJcMw5Du+PMuT7/pSUJttYpiGzJhZe8VNGaahIBdu43OgU3OPzun56vM6880zeu+77yk+GVfg1543wzTkWq72Prk39BJVYYKY3QY6o9QfRHC1F210eo1u2hxG3V7tq9GNqImDbq/br/qDGtNB1yUhAgAAAAwWMzkAYMi++tWv6vHHH9f58+dXXnWrj609/hd/8Rf6jd/4jZVj/Xj/nd/5jsyYKfmSV/VkLVkqXSupcKWg0vWSrEVLTsmRW3XlOZ4C75OZIIZhKGbGFNwiyYH+OvraUb3gv6Bn/+T/Z+/ug+O47zvPf7rnASAHIEGCFIGhBEgy9WDTjCiakuUHWowfopQrzjoiVatIl8hHnS8bZ33Kw25St7teRXXapLJbWznFefImEZNanhSLRZeTPceyubEsR6fYWtGKTzJzkUxbtCGAzwIwMz09/Xh/NAYEIcxMz/MM8H5VdanR3b/uX/+6h6P6fef3+z6oj372o/roZz+qB59/UA8HD7c9BwcAAAAAAOguRnIAQJd96EMfUhAEK+675ZZbKm43DEPvfOc7F7f9xm/8RsvXDzx4QF985otybVel+ZKsC5bMpCkn78hIGPIdX07OkZNz5FoLwQ4nGt0R+IGCMJA2xWkFoHljt45p7Nb2T0kFAAAAAAB6B0EOAOgBprnywLpa25PJy/+Mt2P9mndfo9APVZovqXipqFQmJYWSk3NkJk0FXiC36F4OdBRceUXvcrAjCKRszdsH0GL1JJJmah0AAAAAQD8jyAEAqGg4O6yR60ZUvFiUtcGSmTIVuIHSQ2mZSVNhGMov+XKtaKRHab4kJ78Q7LC8aBTHULfvAgAAAAAAAKsVQQ4AQFU//n/8uP7qE3+ldCYtmVFujnQmLTMdjSYJ3ECe7cnJO1cGOoqu9NNdrjywRjE6AwAAAACwVhDkAABU9Y6D79CJz53Q1DenFIahvKKnVCalRDohwzAU+MHiaI5yoMO6aOna/dfqezd/TyLvOAAAAAAAANpk5cneAQBY4ueO/5yye7OaPT2r3HTu8jKTU+FMQYVzBVkXLFkXLRUuFDTxvgnd/zf3d7vaAAAAAAAAWOUYyQEAiOWBZx7QV37lK/qHv/gH5c/mlRxMKjmQjPJy2L4821NqfUq3f+p2feQ/faTb1QUAAAAAAMAaQJADABDbXb97l/b+i7361u9/S6efOa1L378kSdr8ts26dv+1evdD79bmHZu7XEsAAAAAAACsFQQ5AAB1Gb1pVB/97Ee7XQ0AAAAAAACAnBwAAAAAAAAAAKA/EeQAAAAAAAAAAAB9iSAHAAAAAAAAAADoSwQ5AAAAAAAAAABAXyLIAQAAAAAAAAAA+hJBDgAAAAAAAAAA0JcIcgAAAAAAAAAAgL5EkAMAAAAAAAAAAPQlghwAAAAAAAAAAKAvEeQAAAAAAAAAAAB9iSAHAAAAAAAAAADoSwQ5AAAAAAAAAABAXyLIAQAAAAAAAAAA+lKy2xUAAABAaxmGsbgehmHdx9UqX21/3GsDAAAAANAKjOQAAABYRcpBhnKAYWnQIe5xYRhWDFAYhrG4PwzDK8pV2wcAAAAAQDswkgMAAGCVKQcoagUa4h5X73XrMXNiRtMnpiVJ2b1Zje8ZX/G4yccaq9Pph2qXPf1Q5X2VylYrE+dcnbpmu66xUvk4ZZeX6/T12nnNbrwrK1233uOXa6R8O55Fq9/RRj8/9TyLla7bzLUBAAAQD0EOAAAAtEzc6aqOHjyqk8dOKrU+JRmSFg51LVc779mpg08dbHNNL5t8bOVOyGqdweV9zXSY13tcvdds1zUqla/UjrWuWU2tura6no1es9felXZpdZu2oz1b2S71nKuZdxUAAADNIcgBAACA2OKODpEuT1+11NTzUzp852FltmQ0sW9C6aG0DNNQ4AZy8o7sWVunjp/SowOP6tBzh5S9LSupdodmnA7Eap2p1TohGy3XqFr32oprNnqNSr9qj1u38r56O6KXn7Pd9VzpmsvPG+f4eq/ZiFa1aS2Ntmmj5Zptz0bbpVn1vjcAAABoHjk5AAAAUJeleTfqMfX8lJ742BMa3zOu6z58nbbfvl3ZvVmN7R7T1rdv1eYbNmvkuhGN3jiqzLaMjtx1RG+88Eab7iJSq5O0nsBHJc0GYhq5Zruvsfy4OAGDRjvV6+kAr3Vc3KmY6n3urXpu9b4r3RgdUG+bNlKumfZsVbvU8ywafW8AAADQGgQ5AAAAVpnySItaeTbiHlet/PKRG9UcvvOwNu/YrPE948ruzerqO67W+LvGtfUdW7VpxyaNTI5ow/YNGhob0vD4sFLrUnp83+MN1a3XrIZOzrgduZ3+1fry6/VqPePqxXel0TblWQAAAKATmK4KAABgFSlPJ1UOOFSaPqrWccvXy/ur5dxYPpXV0v1HDx5VZmtGozeOasvNWzS2Z0wjkyPyHV+FcwUlB5MyDEOBF8h3fHm2J9dyZc/b+sK9X9Ddf3l3cw3TJb3aeYu3alXOk2av363r0aF/WSO5OGg/AACA7iHIAQAAsMpUmkZqpaBEPeVr7au2/+Sxk5rYN6HMtoyGtw9rZHJEGyc2yp615Tu+XMuVk3OUHkpHSyatVCalwU2Devmpl9sW5Gg2IXacZMut6vzsRCc4QZnLWhUEiNOmvdBR3gt1iKPd72iz7UDwCAAAoPMIcgAAAKCtZk7MKLU+pfRQWqn1KZkpU77jy5615dmewmBhVEnCkJk0ZaZMmWlTyXQyWgaTOvPSGY3dOtZUPRpJGl2tbLM5NhrViU7TXuiYPf1Q1O4rJZhuZ0d3pXM32/ndSKLzdulke7ZLO9urkXO3670BAABAbeTkAAAAWCVmZmauWJZvP3/+vJ599tnF7Z1anz4xLRmSYRoKg1BuwVXhXEG56ZysC5acvCPP9hR4gcIglMKFabFMyUhEZaZfnG64XdqlWsdwqzuNO9FR2sudseVgR3nphHIC62YSWdcz4qdTaiVy7+WAR7vf0VbceyveGwAAANSHkRwAAACrwCOPPKIbbrhB+/fvf8u+pVNIdWtdoRS4gdy8q+KlopKDSbmWK8Mw5NmeSvMluQVXnu3Jd/0o4OGHUdCjRSp1OFYaLVCrbK1RBq2epmotBjgqJaZud8LqSoGAOO9KWTemNGtG+f56VacCHM2cvxXvDQAAAOpHkAMAAGCVePXVV3Xfffe9ZXs2m11cXxoE6dR6dm90fSfvqDhb1MD5ARmGISfnyEhEycbdgqvSfElOwZFrXRnsMExD2dsu30OrNdoJWatcnJEecafJWosBjqV6uW4rqbdNW/GurGadbAOeBQAAQP8hyAEAAIC2Gt8zLtdyZc/asi5YSg4kFXiB0kNpmUlTYRDKsz05BUeluZKc/EKgw/Lk2dEytru5fBz9iABHdb1a916tV7+iPQEAAFALQQ4AAAC03c57durU8VNRYCMRJR5PD6VlpkwplHzXl2u5cvJRoKM8qsOes7Xr3l3drn5dWjE9US8GOOKOXOl2Z3Sj9WzFdE2NtGmrztWsavfebJs2+s50sg0afRa9Ps0XAADAWkDicQAAALTdwacOyrVc5WZyyp/JKzedU+6NnHLT0d+FswVZ5y0VLxZVvFRUaa4ke86WAunuJ+5ua90qdVDW6rhsV8dmLwY4KpWv9Her1Ho2cYNFtc4X57q91CHfqDjveqfaNO7nq5fbc7lG3hsAAAA0j5EcAAAA6IhDzx3SkbuOKDeVk2u5SmVSSqaTkimFfijf9eVZ3uIIjjAM9eDzD7bs+rU6VevNq1GtXKOWXq/WL+u7cY2lv1pfqWytAECt7Y0+g5Xq0Ug9a5WrpBPPrdo1q21ffs1GA1LtatNaz7ze9mzmXWtUo+8NAAAAWoORHAAAAOiI7G1Z3f/0/cpfyGv29dloNMd0TvmZ/OJojsL5gnIzObl5Vw8cf0Bje9qfi+P0Q5U79Bspt9pV68zu5LXiPJ96trfimr2uWv271aarxWp+bwAAAHodIzkAAADQMdtv367PlD6jYz97TK98/hUlB5MKg1CSZJiGPNvTrnt3xZ6iKk7nYTMdjK3unGy0M7iTdWjHObr1DBot28n7a/Scnbq3bly3lz6z9ZyTYAYAAEB3EOQAAABAxx148oAOPHlAZ146o+kXpyVFIz3Gdrd/5AYAAAAAYPUgyAEAAICuGbt1TGO3EtgAAAAAADSGIAcAAADQx+pJdMx0OgAAAABWGxKPAwAAAAAAAACAvsRIDgAAAKCPMToDAAAAwFpGkAMAgCYZhrG4HoZh3cdVK790X71lAQAAAAAAVjumqwIAoAnlIEM5wLBSUKLWcWEYVgxQlPetdIxhGFfsq3RtAAAAAACA1YqRHAAANKkcfKgVaIh7XCXloEYjAjfQP/zFP+h7X/6ezv6/ZyVJ227Zph0/uUO7P7FbZpLfPQAAAAAAgP5DkAMAgD61PFhSKQDy0p+9pKd/5WnJkFKDKaWH0gqDUK8/87q+95Xv6Su/8hV99Pc/qlseuKVTVQcAAAAAAGgJghwAAPSBlUZxLN+20jFf+sUv6eUnXta2XduU2ZZReigtQ4a8kqfSfEnFN4sqXizqS7/0Jc18e0Y/+dhPduR+AAAAAAAAWoEgBwAAq9QXH/ii/umv/0kT+yY0umNUQ+NDSq5LKnADleZLsi5aGjg/oNRgSmbS1Lf/7NtyLVe6uts1BwAAAAAAiIcgBwAATSqPoKiVZyPuca0w9fdT+sdj/6hr3n+Ntu3apq07t2rD1RtkmEYU4DhvKbkuuZiLI/AD+a6vl/+vl6X/SVK27VUEAAAAAABoGkEOAACaUA5alAMXlaaPqnXc8vVa01AtPefSv8u+/Okva+PERo1Mjmjzjs3a9mPbtHnHZtlztvIzeUmS7/rySp5cy5VTcOQUHJXmSnL/xpX+l8bbpF/EyWdS7bh6ylcqF6c8AAAAAACozOx2BQAA6HdhGC4uy7fHPW75Uu08tc5ZvFjU2ZfPamh8SOtG10XLpnVKrU8pNZhScl1SqXWpaBlMXV5fFyUl1xlJdoON0SeWB5Mqja6pdtxKz6pS+aWqPWsAAAAAAFAfRnIAALDKTJ+YViKVUHJdUolUQmEQyp6zZSQMBW4gv+QrDELJkAzTkJEwZCbNaEmZMhKGwunV3/m+fJRNs8ctF2d6skqjdAAAAAAAQDyM5AAAoAHf+c53dOLECb388suLS1n57yNHjixue+yxxzq2/oXPfUFmwpRCySt5Ks2XVDhbUG46p8L5gkpzJTmWI6/kKfAChUF4xUiFhJGQcb79eUNWs1YFL9489aa++qtf1edu/Zx+e8Nv67c3/LY+t+dz+uqvfVWzr8+2oKYAAAAAAPQ3RnIAANCAL37xi1q3bp1++Zd/+S37brrpJknSxMTE4rYHHnigY+s//vEf11ef/qo825OTc2RdsGQmTTkFR4ZpyC/5cvKOnLwj13Lll3wFbqDAjxY/8KWN8dsCV4ob4Kh13PFfP65v/+m3pVAaHBnUpms3KfADWWctnfgvJ/TS4y9p7y/u1Yd+60OtrD4AAAAAAH2FIAcAAA2yLEvpdPot28vblu4bGRnp2PoNH7hBXwm+otJ8ScVLRaUyKSmUnJwjM2kq8IMo2XjOiZaCI7foyrd9BU4Qdbxn47cD3mr5FFX1juz4rx/5r5o5MaOx3WMa2jakdCZ6l1zbVWm+JPuSLeuCpW999ls6+9JZ3ffl+1pafwAAAAAA+gVBDgAAVpkN12zQ8Niw7Iu2rI2WEqmEAjdQeigtM2UqDEL5JV+u5ao0V1JpvrQ4qsMpOtKwpA3dvov2i5Mzo57jypYHM+oNcBy564jOfOeMJvdPavSGUQ2NDyk5mJTv+CrNlWRdtGQNW0oMJmQkDZ1+7rSe+KknpNtiXwIAAAAAgFWDIAcAAKuMYRp672+8V8f/9XGlhlKSKXmOp3QmCnJIUuAG8oqenIJzRaDDszzpI1Ko1Z0Muxy0KAculgYhlgYlah23fD1uMKNS4OMfj/2jpr45pYl9E7rqnVdp265t2nD1BsmQ7FlbhXMFJQeTMkxDYRBGieQdX69//fUoMHVjXc0AAAAAAEDfI8gBAMAqtPdf7NVLf/aSLr52UWEYyrOjIEcilZAMRbk3FkZzOPko0GFdsLTtlm2aunVKqzzGIalyQGL59rjH1XOdSmWf+ffPaNO1m7RxYqM2v22ztr5zq0ZvHFXxYlG56Vw0Csfx5Rbd6NkVotwqpUxJ7t+6BDkAAAAAAGuO2e0KAACA9vjkC5/Uhu0b9OYP3lTujVy0zOSUP5NX4UxBhXMFWRcsWRct5c/ltWnHJj349w92u9prVn4mr0unLmn9Veu1btM6DYwMLObiMJOmEgMJJQeTb13WJaO8KxcloxBvSi0AAAAAAFYLRnIAALBaGdKnvvspHb3nqL7/37+vwrmCUutSSgwkpDBKYu0VPZlpUzf/s5t14MkD3a7xmjZ9YnoxmGEkDAVeIHvWjkbeeIE821PgBVIYTXdlmIbMhCkzGS1GwlA4vQaG4AAAAAAAsAQjOQAAWOXuOXqP7v2re3XdB6+TkTCUm45GdCTSCV3/4et133+7b80EOJ599lmdOHHiiqXstdde0yOPPLL4d6fXZ78/KzNxOTF8aa6k/Jm8ctM5WecslWZLcguuPNuT7/oK/VBhEC4GPRJGQrrUULOsGuX8KXGSyVc6rtF99VwfAAAAANA6jOQAAGANmPzApCY/MCkpSjouaTEJ+Vry9a9/XWEY6jd/8zffsu+GG27Qww8/vPh3p9df/b9fjUZsFD2V5qMcKWbClJN3ZCZM+Y4vJx/l4HCty8GOwAsUeIH8wJc219Maq8vS5O/lQMNKuU+qHbe8TNx9K/0NAAAAAOgMghwAAKwxazG40Q+y78oq8AOV5ksqXioqtT6lMAiVzqVlJk2Ffii36MrJOdFScORZXhTscHyFQagwu7Y72ctBhnIAo97jqgUpqu1rJMAxc2JG0yemJUnZvVmN7xlf8bjJx+o67aLTD9Uue/qhyvsqla1Wplq5uOVXOkecMnE1el/Vyrfznlp1vXZes9k2rXSuet+1dr+bnXoWrX5HG/38NPMsmrkuAABAIwhyAAAA9ICh8SGN7hhV8UJRhY0FmUlTvusrPZRWIpWIprFyfDkFR878wrIwqsMtutKopEy372LtihssOXrwqE4eO6nU+pRkSFo41LVc7bxnpw4+dbDNNb1s8rGVOyKrdQaX97WjA7PRgE4rzh/nviqVr9SOta5ZTa26trqejV6zle9K3LZq93tSzzVb/Syaac9WtksrnkU7/60AAABYjiAHAABAj/jgb31Qx+47ptRwSoZhyCt5i0EOKZpqzLVdOXlHpfmS7HlbTt6RV/Skn+ly5VeZaqMzVtpXa+qqqeendPjOw8psyWhi34TSQ2kZpqHADeTkHdmztk4dP6VHBx7VoecOKXtbVlLtDs04HYjVOlOrddI2Wi5uvWqVbUdndq32jNP5vHR/ve1R7z0tP2e767nSNZefN87x9V6zEa1q01oabdNGyzXbnu38/KxUn6XXXGl/u54/AADAUsxXAQAA0CNu+umbdO2d12ru9JxyM7koSfwbucVk8fmzeVnnLRUvFmW/aas0W5J10dL1H75euqHbtV896g1w1DL1/JSe+NgTGt8zrus+fJ22375d2b1Zje0e09a3b9XmGzZr5LoRjd44qsy2jI7cdURvvPBGK26lolqdpPUEPlql2nVbdf5G9lU7Lk7AoNFO9Xo6wGsdF3e6oXqfe6sCXfUG7brRaV5vmzZSrpn2bFW71DsCoxv/VgAAACxHkAMAAKCH3Pel+zTx/gnN/XBO+el8FOCYzik/k1f+bF6FswUVzhdkXbBUuFDQ2z70Nt37V/d2u9o9oTxlVLV8HLWOa3WAQ5IO33lYm3ds1viecWX3ZnX1HVdr/F3j2vqOrdq0Y5NGJke0YfsGDY0NaXh8WKl1KT2+7/G6r4P2iBsA6PQ0Ssuv16v1jKsXO8UbbVOeBQAAQGcxXRUAAECPuf/L9+tv/+3f6sU/fFH5s3klBhJKDkT/2+bZnvySr8S6hN73r9+n/Y/s725le0Q5iXg5cFFp+qhaxy3979L91fZVS2B+9OBRZbZmNHrjqLbcvEVje8Y0Mjki3/FVOFdQcjApwzAUeIF8x5dne3ItV/a8rS/c+wXd/Zd3t6B1Oq+dycPRWt3OndDt4BDv5mW9GnQBAACohSAHAABAD/rQf/iQ9v6ve/XCH76g1//2dV187aIkafONm3X9h67X7f/ydm24ekOXa9lbKo2yqJY/I872Wvuq7T957KQm9k0osy2j4e3DGpkc0caJjbJnbfmOL9dy5eQcpYfS0ZJJK5VJaXDToF5+6uW2BTmaTYjdyimYegGdu5e1KggQp0174b3ohTrE0akcG/VMUzX5WO2cJL3ergAAYHUgyAEAANCjNk5u1Ed+5yPdrgYaNHNiRqn1KaWH0kqtT8lMmfIdX/asLc/2FAYLo0oShsykKTNlykybSqaT0TKY1JmXzmjs1rGm6tFI0uhqZetNXN1PAYRe6JCt1nnczrasdO5mO6sbSXTeLv38bpZ1K29NpePL72orzgcAANAocnIAAACgr83MzFyxVNr+3e9+d3Hfs88+2/b16RPTkiEZpqEwCOUWXBXOFZSbzsm6YMnJO/JsT4EXKAxCKVyYDsuUjERUZvrF6Waapi3qDZos3daLncq9/IvzcgdytY7kVisnsG4mkXWcNu30u9CP72ZZu9/RXr53AACAOBjJAQAAgL71yCOP6IYbbtD+/fvfsq/aNFUdWw+lwA3k5l0VLxWVHEzKtVwZhiHP9lSaL8ktuFGuFdePAh5+GAU9WqRSx2i1qWaqlY1TbqXz9GJHaq8GOColpm53wupKgYB6nnk9AY5eaPdefTfLOhXgaOT81co28u8EAABAowhyAAAAoK+9+uqruu+++96yPZvNVvx7aVCkXeszG6JRJU7eUXG2qIHzAzIMQ07OkZGIko27BVel+ZKcgiPXujLYYZiGsrddeQ+tVG/ndbPlek0vdbRX0st1W0m9bVotuNAPz6fdOtkG9T6LWnVbLf9OAACA/kCQAwAAAGiD8T3jci1X9qwt64Kl5EBSgRcoPZSWmTQVBqE825NTcFSaK8nJLwQ6LE+eHS1ju5vLx4GV9XMHeq/WvVfr1a9oTwAAgPgIcgAAAABtsvOenTp1/FQU2EhEicfTQ2mZKVMKJd/15VqunHwU6CiP6rDnbO26d1e3q98yvTQdUL2dx7V+kd4rndGN1rMV0zU10qatOlezauWYaaZNG31nOtkGvfQsAAAAGkXicQAAAKBNDj51UK7lKjeTU/5MXrnpnHJv5JSbjv4unC3IOm+peLGo4qWiSnMl2XO2FEh3P3F3W+tWqXO3Vod3veWWbu92Z2mznbbL77FdwZtabVmr/o3Wc6XjeqlDvlGteDdb1aZxP1+93J5LNfrvCAAAQCsxkgMAAABoo0PPHdKRu44oN5WTa7lKZVJKppOSKYV+KN/15Vne4giOMAz14PMPtuz6tTobq/3SvB3lKonbWdqKAEWtX++vtK1cZqWytQIAtba3qi0brWetcpU006aNarRNG30329WmtZ55ve3Z7s/PSnWI8870S7AGAAD0N0ZyAAAAAG2UvS2r+5++X/kLec2+PhuN5pjOKT+TXxzNUThfUG4mJzfv6oHjD2hsT/tzcZx+qHKHfqvLVSrTT6p1ZnfyWnF6UkrIAAAgAElEQVSeTz3bW3HNXtfsu9mONl0NqrXdanhvAABA/2AkBwAAANBm22/frs+UPqNjP3tMr3z+FSUHkwqDUJJkmIY829Oue3fFnqIqTudhMx2MjZbtxjU7ef56z9Gt9ujU8+tkgKfZazZb105ct9c+P63IsQIAANAJBDkAAACADjnw5AEdePKAzrx0RtMvTkuKRnqM7W7/yA0AAAAAWI0IcgAAAAAdNnbrmMZuJbABAAAAAM0iyAEAALDKGIaxuB6GYUPHtXpf3DoBcdWTSJrpdAAAAIDVi8TjAAAAq0g5mFAOJCwNLsQ9rta+MAwXl7j7ytsAAAAAAGglRnIAAACsMuVgwvJAQz3HxT1HO82cmNH0iYW8FXuzGt8z3pV6oDcxOgMAAACARJADAAAAdVhphEacffU4evCoTh47qdT6lGRIWjiNa7naec9OHXzqYEPnBQAAAACsPgQ5AAAAEFt5SqqV/q62L46p56d0+M7DymzJaGLfhNJDaRmmocAN5OQd2bO2Th0/pUcHHtWh5w4pe1u2dTcGAAAAAOhLBDkAAADQdVPPT+mJjz2h8T3jGr1xVJltGaXWpxQGody8q+JsUdYFS+mhtHIzOR2564juf/r+blcbAAAAANBlBDkAAABWmfIIilq5NKodF/ccrXL4zsMa3zOu8T3j2nLzFg1vH5aZMuUWXBUvFTVwfkDJgaTMhClJyk3l9Pi+x6V/05HqAQAAAAB6FEEOAACAVaQcmCgHJypNH1XtuDj7lv4dZ9/S7cvPe/TgUWW2ZjR646i23LxFY3vGNDI5It/xVThXUHIwKcMwFHiBfMeXZ3tyLVf2vC3nqCORogMAAAAA1iyCHAAAAKtMpTwYy7dXy5fR6n3Vypw8dlIT+yaU2ZbR8PZhjUyOaOPERtmztnzHl2u5cnKO0kPpaMmklcqkNLhpUM5Jp+J5AQAAAACrn9ntCgAAAGDtmjkxo9T6lNJDaaXWp2SmTPmOL3vWlmd7CoOFUSUJQ2bSlJkyZaZNJdNJJdNJKSkZZzozpRYAAAAAoPcQ5AAAAFglbrrpJs3MzCwu3/jGNxb3Pfvssz25Pn1iWjIkwzSiJOMFV4VzBeWmc7IuWHLyjjzbU+AFCoNQChemuzIVBT4MU5qO0zoAAAAAgNWI6aoAAABWgYcffljT09NvyZHRD+sKpcAN5OajJOPJwaRcy5VhGPJsT6X5ktyCK8/25Lt+FPDwwyjoAQAAAABY0whyAAAArBLZbLbi3/v37+/J9ezeqI5O3lFxtqiB8wMyDENOzpGRiJKNuwVXpfmSnIIj11oW7AhDhVmCHQAAAACwVhHkAAAAQNeM7xmXa7myZ21ZFywlB5IKvEDpobTMpKkwCOXZnpyCo9JcSU5+IdBheVHODi+Uxrp9FwAAAACAbiHIAQAAgK7aec9OnTp+KgpsJKLE4+mhtMyUKYWS7/pyLVdOPgp0lEd12HO2tLPbtQcAAAAAdBNBDgAAAHTVwacO6tGBR5WbyUmSPNtTOpOWmTZlGNGUVZ7tybWiaatKc6UowBFIOiiJ2aoAAAAAYM0yu10BAAAA4NBzh+TmXeWmcspN5zQ/Pa/8dF65mZzyZ/IqnC/IOm+peKko65KlMAz14PMPdrvaAAAAAIAuI8gBAACArsveltX9T9+v/IW8Zl+fVW46CnbkZ/JRkONsQYXzBeVmcnLzrh44/oDG9pCMAwAAAADWOqarAgAAQE/Yfvt2fab0GR372WN65fOvKDmYVBhEc1EZpiHP9rTr3l26+4m7u1xTAAAAAECvIMgBAACAnnLgyQM68OQBnXnpjKZfnJYUjfQY283IDQAAAADAlQhyAAAAoCeN3TqmsVsJbAAAAAAAKiPIAQAAgFXBMIzF9TAMGzquHfsAAAAAAO1D4nEAAAD0vXKQoRxgWBp0iHucYRgKw3BxacU+AAAAAEB7MZIDAAAAq0I5cFEr0FDpuE6OwJg5MaPpEwv5RvZmNb5nfMXjJh9r7PynH6pd9vRDlfdVKlutTJxz1Vu+mbK1zrdU3HOvVD5O2eXlOn29dl6zW+9KvW3azOehUvl2PItWv6ONfm5qPYu4/za14nMLAABQC0EOAAAAoEOOHjyqk8dOKrU+JRmSFuIqruVq5z07dfCpgx2ry+Rj9Xdelvc102EeVzNl6z1fnPuqVL5SO9a6ZjW16trqejZ6zW68K61+L5q5ZqufRTPt2cp26UYbAwAANIMgBwAAALBMeQqqsmqjQ+JMUTX1/JQO33lYmS0ZTeybUHooLcM0FLiBnLwje9bWqeOn9OjAozr03CFlb8tKqt2hGacDuVpnarVO2kbL9bJa7Rmn83np/rjtUd5Xb+fx8nO2u54rXXP5eeMcX+81G9GqNq2l0TZttFyz7dlou9QrTnCnH/+NAAAA/YmcHAAAoCLDMBaXRo9rdF891wdaaXmAo2xp3o169k09P6UnPvaExveM67oPX6ftt29Xdm9WY7vHtPXtW7X5hs0auW5EozeOKrMtoyN3HdEbL7zRlnsrq9VJWk/go5JmOjrb0Ukap2O43nPECRg02qleTwd4rePiTsVU73NvRZvGuf7y83aj87zeNm2kXDPt2ap2IUABAAD6EUEOAACwom4mcl5pP1BL+R2KE5SrdFylAEfcY1bad/jOw9q8Y7PG94wruzerq++4WuPvGtfWd2zVph2bNDI5og3bN2hobEjD48NKrUvp8X2PV61Dv2imo7RXOlnjBgA6PcXP8uv1aj3j6pXnvVSjbbqWn0Wv3hMAAFjdmK4KAABU1M5EztX2xeloXsnFVy9KkkZvHK27LPpb+d1bHnSTrnyfah239L9L91d7r6vtO3rwqDJbMxq9cVRbbt6isT1jGpkcke/4KpwrKDmYlGEYCrxAvuPLsz25lit73tYX7v2C7v7LuxtvlC7qpVwcqK7bv9zvdnCoF4Mr3dLKZ0G7AgCATiLIAQAA2ipu0KPRci/9yUv6+9/9e1383kUl09H/2niOp9Edo3rvv3qvdh/a3UCt0Y8qvSfLt8c9rhX7Th47qYl9E8psy2h4+7BGJke0cWKj7FlbvuPLtVw5OUfpoXS0ZNJKZVIa3DSol596uW1BjmYTYrdrPv5udbgTWLmsVUGAOG3a7QBLr9Qhjna/o/3SDgAAACshyAEAANqq0i/ql1ppe61yXsnTn777TzX3ozmt27RO2/dul5k2FxM5F84X9JVf/Ype+IMX9MkXPikjQV4PdNbMiRml1qeUHkortT4lM2XKd3zZs7Y821MYLIwqSRgyk6bMlCkzbSqZTkbLYFJnXjqjsVvHmqpHI0mjq5VtNNl5XN3sZO2FDt7TD0XtvlKC6XZ2dFc6d7Od340kOm+XTrZnu7SzvZo9N4ESAADQLeTkAAAAXdXI1FSlXEm/d93vqTRb0uT7J3X9h67X9ju2a2z3mK5651XacvMWXfWOq7Tx2o168wdv6ncnfldOwWnTHaDbbrrpJs3MzCwuZUu3Ld/+5ptv6tlnn13c1o71rz/5dcmQDNNQGIRyC64K5wrKTedkXbDk5B15tqfACxQGoRQujGAyJSMRlZl+cbqJlmmPah3D/ThNVS93zJaDHeWlE8oJrJtJZF3PiJ9OqZXIvZcDHu1+R3v53gEAAOJgJAcAAKioHICIk8i5WvLwWuXq9Ufv/COlMildfcfVGr1pVMPZYSXSCXm2J3vWlnXBin45nzZlGIbmfjSnP971x9IDdV8KPe7hhx/W9PT0iu9RrWmplu5vx3q0QQrcQG7eVfFSUcnBpFzLlWEY8mxPpfmS3IIrz/bku34U8PDDKOjRIpU6RiuNFqhVttYog36cpqrXAhyVElO3O2F1pUBAnHelrN1TmrVa+f56VacCHL3wLAAAABpFkAMAAKyoFYmcqyUir5bkuVq5r/27r6k0X9L1H75eW3du1dgtY9pwzQaFQajixaLy5/Iyk2bUuVxO5lzyNHd6Tvq6pDubbRn0mmw229D2/fv3L25ry/p9+/WDP/qBnLyj4mxRA+cHZBiGnJwjIxElG3cLrkrzJTkFR651ZbDDMA1lb1v5Hlqh3s7ruOXijPSoFnhptGy9+qFzt5frtpJ627STz7sfdbINmn0WPC8AANBNBDkAAEBFzSZyrucccfYHXqAX//hFjd44upjEefSmUW26fpOKl4oyTCMKalie3EKU0LmUKyk9lNbAxgG5L7gyPkBuDnTG+J5xuZa7OLooOZBU4AVKD6VlJk2FQSjP9uQUHJXmSnLyC4EOy5NnR8vY7ubycWBl/dwh26t179V69SvaEwAAID6CHAAAoG+8+f035VmeBjcNamB4QKn1KRmGIddyoyTOphElb06ZSqQTSgwklBxIKjmQVGpdSroghW+2bhogoJad9+zUqeOnosBGIko8nh5Ky0xFo41815druXLyUaCjPKrDnrO1695d3a5+XZr5lXcnfyFe7/nijlzpdmd0o/VsxXRNjbRpq87VrGr33mybNvrOdLINWvEsenm6LwAAsDaQeBwAALzF6Oiocrnc4lK2dNtK2/P5vE6fPr24vdXrMydmJENKpBKSJK/kybpoKTedU/FiUW7Ble/4CoMwGgmykPTZSBgyk6ZMw5R6L48zVrGDTx2Ua7nKzeSUP5NXbjqn3Bs55aajvwtnC7LOWypeLKp4qajSXEn2nC0F0t1P3N3WulXqmKzVYdnPHZrNdh4vv/d2tUWtZ1Nvp3Pceq50XC91yDcqzrveqTaN+/nq5faspB/rDAAAVgdGcgAAgCt84hOf0NzcnGZmZha3DQ8PS9IV21bankgk9Hd/93f6xCc+IUl65plnWrpuXbBkmAu5DIqu7Ddt5c/kFxM5u8Voiiqv6Ml3fAVuoMAPpCCa/sqUqcAKWthaQG2HnjukI3cdUW4qJ9dylcqklEwnJVMK/VC+G02xVh7BEYahHnz+wZZdv1anar15NaqV62VL76nWr/dX2lYus1LZenOMVEooHqdsNY3Ws1a5Sppp00Y12qaNBqTa1aa1nnm97dnMuwYAANDvCHIAAIArTE5OVtx344031tz+tre9bXG9HKRo1fq2H9umMAijHAazJRXOF2SYhpy8szgVkFNwVJovyS0sJHF2fPmur9AL5Ye+wm1MV4XOyt6W1f1P36/H9z0ue97W4KZBJdNJGQlDYRAq8AJ5trc4guPB5x/U2J725+KI0zlbT7nVrlK7tKM9mrlWo2U7eX+dVu2dpk2b088jTwAAwOpBkAMAAPSN8XeNy3d8leZKsi5aSq5LKgxCpXMLiZz9KJFzKbeQ26CcyNn25JU8hX4oYzuJx9F522/frs+UPqNjP3tMr3z+FSUHo3dXiqZU82xPu+7dFXuKqrgds41qdYdlL9SlFeep9xzduu9Gy3by/ho9Z6furRvX7YXPSb3nJLgBAAB6AUEOAADQN9JDaV3znmt08bWLGhgekJk05bu+0pkliZydKJFzKVdSaa4kJ+fIKThy8o40IYVJRnKgew48eUAHnjygMy+d0fSLUYKY7G1Zje1u/8gNAAAAAFiNCHIAAIC+8jNHfka/f+PvK38uLxlR8vF0Jq1EOkpGXp76pzxtVTnQ4dmedKDLlQcWjN06prFbCWwAAAAAQLMIcgAAgL6ycWKjPvDwB/Tcbz2nMAjlFt3LQQ5jIZGz40dJyPMLgY5cSR/8Dx/U8cJxiYEcwKpQTyJpptQBAAAAVi+CHAAAoO/s+9/3KTWY0tf+7dfk5BwNDA8oMZCQYRoKw1CBuzCaI+fItV39xH/8Cd32L2/T8UeOd7vqAAAAAACghQhyAACAvnTHr9yhifdN6Og/P6r8mbx8z1ciFU1Z5TvRemZLRj//339eY3uYFghYbRidAQAAAEAiyAEAAPpY9vasHvr+Q/rh//ND/eBvf6AfPf8jSdI177tG133wOk28f6LLNQQAAAAAAO1EkAMAAPQ3Q5p4/wQBDQAAAAAA1iCz2xUAAAAAAAAAAABoBEEOAAAAAAAAAADQlwhyAAAAAAAAAACAvkSQAwAAAAAAAAAA9CWCHAAAAAAAAAAAoC8R5AAAAAAAAAAAAH2JIAcAAAAAAAAAAOhLyW5XAACwNhiGsbgehmHdx9Uq3+x+AAAAAAAA9B9GcgAA2q4cYCgHF5YGHOIeF4ZhxeCEYRiL+8MwfMv5l+8HAAAAAADA6sBIDgBAR5SDCysFIRo5bqUyKykHOOI6+52z+uHf/VCnnzstSZrcN6mJ909o2y3bYp8DAAAAAAAAnUGQAwCw6sWZqurCP13Q5z/+ec1NzckIDSXXJ6VAevWvX5US0sarN+q+/3afNu3Y1KlqAwAAAAAAoAaCHACAVWWlkRvLc3ss3//t//JtPf3LT2vj5EZNvG9C6aG0DMOQ7/gq5UqyZ23lz+T1B7v+QD/1hz+l3f/z7o7cCwAAAAAAAKojyAEAWDXqnZpKkv7HH/wPfe3ffU1Xv+dqbd6xWUNjQ0quSyrwAjl5R8VLRVkXLKXWp5Q+m9aXH/qyfMdv0x0AAAAAAACgHgQ5AAAdUQ5A1MqzEfe4SuXqUThf0Fd+9Su65r3XaNuPbdPWd2zV8PZhmQlTTt6RddFSOpNWIp2Izh+ECrxAf/Ppv5F+VdJgXZcDAAAAAABAixHkAAC0XTloUQ5cVJo+qtZxy9fL+8t/r5R7Y3nAZOk5j/3zYxq+elgj141o9KZRbdu9TZuu2yTXcpU/k5eZMhUGoXzXl2d7couuXMtV+s207KO29HMtaBwAAAAAAAA0zOx2BQAAa0MYhovL8u1xj1u+xNlX6Zxe0dPr33hdI9eMKLM1o8xVGQ2PDWv9lvUa2DCg9HBaqUwqWtZfuQxsHJB+IIlZqwAAAAAAALqKkRxY0yr9ujvucbXKxykX5/oAWm/6xLSSg0mlMiklB5MyDENOwZF13pJX8hS4gRRGn1czYcpMmkqkEtGSTshIGtJ0t+8CqF8rv/uqnWP5FHJ89wEAAAAA2oGRHFizKk13U89xK/1ifGm5pb8eX6lctfLAajA2Nqb3v//9OnXq1OJS9vnPf76r62e/c1amYUqG5Lu+nLyjwrmC5qfnZV2wVJovySt68h1fgR9IgaRQkiEZpiHTNKUz9bYI0F2t+O4rb6/1Hbgc330AAAAAgHZgJAfWtEpz9jd63EplamkkWTLQL37hF35Bs7OzK+675ZZburo+sHFAQRDIL/mLScYT6YScgiPDMOSXfJVyJbkFV17Rk+d48j1foR8qDBY6aUk8jj7Uzu8+6fL3WrUycb/7Zk7MaPpENGQquzer8T3jKx43+Vjs6l3h9EO1y55+qPZ5VjpHtXKtuGYcla4T9/z13lelcp2+Xruu2Wx7VjtfrXPU26bNvmOdehatfkfb/a5VO0+rPrcAAACoH0EOoI3iTglSzdS3pvSt//Nbmvr7Kc39aE6StGFig655zzV67796b8UOH6BXjIyMrLj95ptv7up69l1ZhWGoUq4k+01bhXMFSVI6l5aZNBV4gVzLVWm+JCfvyLVcebYnr7QwuiMMpGyMBgBWoWpTWbUicH/04FGdPHZSqfUpyVA0ikqSa7naec9OHXzqYNPXiGvysfo7vZfu61bHZ7N1q1S+0faoplZdW13PRq7Z6mcdt60abdNmtLpNK5Vrpk3b9a5Vu2ar6gAAAIDWI8gBtFGtjp9anUFffOCLeu1Lr0mmtG7zOmXflVXgBSpeKuq1v3lN33v6e3r7z7xdP/1nP922ewBWq9EbR5VIJmS/acs6H43iCLxA6aG0EqmEwiCUZ3tyCk4U6Mg5cvMLozpsTzKlcDOjsLD2VPouixvgqHbc1PNTOnznYWW2ZDSxb0LpobQM01DgBnLyjuxZW6eOn9KjA4/q0HOHlL0tijTW6giN02lZrRO2Vqd3vWWrlWulWu0Sp/N56f4497S0TL2dwJXauF31XOmay89b69h6r9eoVrVpLY22aaPlmm3TetqlFZ9ZAAAA9CZycgA96k/2/ole+9JrGrt1TDvu2qFr91+rsT1jumrXVRq7dUxjt4xpeGxY333qu/rTO/6029UF+o6RMLTnk3uUm8mpcL6g/Exeuenc4pI/k1fhfEHFC0UVLxVlz9oq5aJgRylXkvYq+oU5gEWGYSwu5b/jmnp+Sk987AmN7xnXdR++Tttv367s3qzGdo9p69u3avMNmzVy3YhGbxxVZltGR+46ojdeeKNdtyKp8eBI3LLtFKdjuN5zxAkYNNqpXk8HeK3j4k7FVM+za0V7xr3+8nN3412qt00bKddMmzbTLq34zHZ7pBYAAACuRJADa1rcTphGOmvqOXa5x9/3uOam5nTtj1+r7bdv1/Z3Rx0923Zt0+hNoxq5bkQbJzZqePuwRq4d0fnvntef3/nnDV8PWKt+4j//hNKZtOZ/NK/cmZzy00sCHTM5Fc4UokDHxaKKb0aBjuKbRa3fvF76SLdrDzSmXd99KyUWr2fqqsN3HtbmHZs1vmdc2b1ZXX3H1Rp/17i2vmOrNu3YpJHJEW3YvkFDY0MaHh9Wal1Kj+97PPb5EU/cAECnp+pZfr1erWdcvdg53mib9vuzaFQvPkMAAIC1iumqsGaVp9Yod95Umlqq1nHL1yslao07VdV3/uI7OvfKOV27/1pd9c6rdNWuq7Thmg2StJg3IJFORNN3+IECL5Dv+Jp+cVraKumdDTcJsCZ96pVP6Q/f+YeaOz0nz/KUyqQWP2NhEMp3fHnFaNqq4ptFpYfS+sVXflG/89jvdLvqQN1a/d23fF8tlb77jh48qszWjEZvHNWWm7dobM+YRiZH5Du+CucKSg4mZRjG4neeZ3tyLVf2vK0v3PsF3f2Xd8dvhB7SaNJkNK7bv8DvdnCId6x5qy1YAwAAsBowkgNr2vJfnC7dHve4lX65WqvcStco+8aj39Dmt23Whms2aOT6EV218yptv227Nl27SUPbhrR+dL3WbVqnwY2DGtw4qIHhAaWH0xoYHpC+3kAjAGvcutF1+qX/75e0bnSdcm/kNHd6bnEkR/5MXoVzBeVmcipeLGrD9g369Kuf1uDIYLerDTSs1d991a4TZ5sknTx2Upt2bFJmWyYapTgZjVhcv2W9BkcGNbBhQOmh9OUlk1Yqk9LgpkG9/NTLcW+9btU6M2v9Or3RZNSd6EClk/aycps30/b1JhHvZqChU+9Ys9pRx1Z8ZnvhGQIAAOCtGMkB9JD5H81rbmpOk/smNTgyqMENg0quS8p3fRkJQ2baVGIgsbgkB5JKDCaUGkwpuT4p/UhSrtt3AfSfwY2D+vSrn9Y3//M39c3f+6bmfjin9FBakuTkHW2c3Kj3PPQevftX3t3lmgKrz8yJGaXWp5QeSiu1PiUzZcp3fNmztjzbUxgsjCpJGDKTpsyUKTNtKplORstgUmdeOqOxW8eaqkecgMZK26t1GteTc6AbHc+90FG7tA072SatDE6VNZPDpR164R1rVqvbq5nPbLvqBAAAgOYR5MCa5HneFX8nk9FHoVQqaWBgQJJULBa1bt26jq5Pn5iWmTAXp8rxXV/FS8Xo17JeKK/oKXCDy1NhmZKZMGUkDSWSCZmmqWA6aE+jAWvAHb92h+74tTukUNEUcJKyt2W7XCugNWZmZq74e3x8vOXbX3vtNX3gAx+QJD377LO68847a64P/dOQZGhxiji34KpwriDf8RUGoZy8I8/2FHiBwiCUQi1+BxqJqMz0i9NNBzk6qVJi63LH60od/q3Sy79E74VAT711iNuenb63br5jzerVd7Qfg0QAAABrBdNVYc358pe/rJdeeknf/e53F5eyr371q4vrn/3sZzu+Pn96XmbCVBiE8mxP9qyt/Jm88tPRlDn2rC2n4MgrelHnjx8udvhIkmmY0mx97QFgBUYU3CDAgdXikUce0TPPPLPiNFOVpp9qdPvSv2Ovh1LgBnLzroqXisqfySs3nVPhbEHFi0WV5ktyC64825Pv+lHAo/wd2CKnH1p5keL96r+ectXq0E692nm8tM3ibG/ldStti/Ps6g1w9EK790Idqml3WzX6me2lZwgAAIC3YiQH1pwXXnhBYRjqN3/zN9+y72Mf+9ji+q//+q93fH3zTZsVeIFcy1VpviTrgiUzYcopODLNaPoOp+DIyTtyrYXOHsdX4AYK/EB+4EtbYjcFAGANefXVV3Xfffe9ZXs2u3Iwr5HtS/ft378/1vrMhmhkiJN3VJwtauD8gAzDkJNzZCSiZONuIfpedApLvv8Wgh2GabQ1IFlpOqVanZ7VpmHqhn7opO3lui3XaM6VVp5vtelkgGMlcT6zPEMAAIDeRJAD6CHZd2UVeIFK8yUVLxaVWp9SGIRK59Iyk6ZCP5RbdOXknKizJ+/ILbrySt7itB5htnW/agUAoN3G94zLtVzZs7asC5aSA0kFXqD00MJ338LoRqfgqDRXuhzotzx5drSM7e6fqaq6oZ87X3ux7r1Yp35HmwIAAKAZBDmAHrJ+63pdtfMqWectFTYUFpOvpofSSqQSCsNQfslfHOlRDnQ4hWjRVZLWdfsuAACoz857durU8VNRYCNx+bvPTJlSKPlu9N3n5KNAR3lUhz1na9e9u7pd/ZZpx5z/9XYe1/o1e690Rjdaz6V5KRrRyP1XO7bT7Vnt3ptt00bfmV55p6rppWcIAACAtyInB9Bj7vrdu2RdslQ4V1B+JsrHkZvOKTedU34mys1hXbBUvFSUPWtHv2rNOfJLvvTRbtceAID6HXzqoFzLVW4mt5iPI/fGwnffmbwKZwuyzlsqXiyqeKmo0lxJ9pwtBdLdT9zd1rrV6hCvNYd/I9tb1VnabOfr8rq2K/Fys3kQGq3nSsdVu2a/dGa34h1rVZvG/fx0sk3r/WwCAACg9zGSA+gxk3dO6qZ/dpNe/etXoyk6ip5SmZQS6YQMw1AQBIujOZx8NG2V/aatd9zzDn3nmu8sJhkfT3AAACAASURBVCEHAKCfHHrukI7cdUS5qZxcy1Uqk1IynZRMKfRD+a4vz/IWR3CEYagHn3+wZdev1cFZbURArV/H13utVlh6jXrrV+ve6s1XsHx7q9qk0XrGfXaV6ldvezaj0TZt9B1rV5vWeuat+gxVa5dmPrMAAADobYzkAHrQgScO6O0H367cVG5xFEduOqfcmSW/aL1gyboYjejYdd8uffwvPt7tagMA0LDsbVnd//T9yl/Ia/b12StGMZa/+wrnC8rN5OTmXT1w/AGN7Wl/Lo7TD1Xv2K13X7UO1Grn67Rq99XJa9W6XqP1bOaava7Zd6wdbdorGv08AwAAoLcxkgPoUR//849rfM+4vv7vvy7rgiUjYSg5kFSoKC9H4AVKpBP6yH/6iG771G3dri4AAE3bfvt2fab0GR372WN65fOvKDmYVBhEQxQN05Bne9p1767YU1TF6bBsRadmvefoREdqv91XN8q2Kq9GMxoNHDR73naV71Sbduv+2n0uAAAANIYgB9DD3v2/vVu7f363vv1n39apr57Sue+ekyRl92a14yd2aM8n9yg9nO5yLQEAaK0DTx7QgScP6MxLZzT94rSkaKTH2O72j9wAAAAAAPQXghxAjxsYGdB7fu09es+vvafbVQEAoKPGbh3T2K0ENgAAAAAAlRHkAAAAa5JhGIvrYRg2dFwj+5Zur3Z9wzCq1gtrSz2JpJk+BwAAAMBaQuJxAACw5pQDDeUgwkqBh1rH1doXhuHisjzgsXSpdl0AAAAAAFAdIzkAAMCaVA4wLA9C1HNc3HNUstJojfK2uOebOTGj6RMLeSv2ZjW+Z7zueqD3MToDAAAAAFZGkAMAAKBH1DNF1dGDR3Xy2Eml1qckQ9JCMddytfOenTr41MH2VRQAAAAAgB5BkAMAAKDF4ozEWB7QiBvgmHp+SofvPKzMlowm9k0oPZSWYRoK3EBO3pE9a+vU8VN6dOBRHXrukLK3ZZu+HwAAAAAAehVBDgAAgDaolWx8JcuPWx74mHp+Sk987AmN7xnX6I2jymzLKLU+pTAI5eZdFWeLsi5YSg+llZvJ6chdR3T/0/e35oYAAAAAAOhBBDkAAMCaFDf3RbXj4pwj7giNSrk5ljp852GN7xnX+J5xbbl5i4a3D8tMmXILroqXiho4P6DkQFJmwpQk5aZyenzf49K/qXl5AAAAAAD6EkEOAACw5pQDE+XgRKVpo6odV+scS49brp7cG2VHDx5VZmtGozeOasvNWzS2Z0wjkyPyHV+FcwUlB5MyDEOBF8h3fHm2J9dyZc/bco46Eik6AAAAAACrkNntCgAAAHRDGIaLy/LtcY6rdY5KZVa6RpxjTh47qU07NimzLaPh7cMamRzRxomNWr9lvQZHBjWwYUDpofTlJZNWKpPS4KZB6WTNywEAAAAA0JcIcgAAAPS4mRMzSq1PKT2UVmp9SmbKlO/4smdtebanMFgYVZIwZCZNmSlTZtpUMp1UMp2UkpJxJl5eEAAAAAAA+glBDgAAsKbcdNNNmpmZWVy+//3vL+579tlne3J9+sS0ZEiGaURJxguuCucKyk3nZF2w5OQdebanwAsUBqEULkyZZSoKfBimNB2ndQAAAAAA6C/k5AAAAGvGww8/rOnp6bfk1uiHdYVS4AZy81GS8eRgUq7lyjAMeban0nxJbsGVZ3vyXT8KePhhFPQAAAAAAGCVIsgBAADWlGw2W3Hf/v37e3I9uzeqs5N3VJwtauD8gAzDkJNzZCSiZONuwVVpviSn4Mi1lgU7wlBhlmAHAAAAAGD1IcgBAADQ48b3jMu1XNmztqwLlpIDSQVeoPRQWmbSVBiE8mxPTsFRaa4kJ78Q6LC8KGeHF0pj3b4LAAAAAABajyAHAABAH9h5z06dOn4qCmwkosTj6aG0zJQphZLv+nItV04+CnSUR3XYc7a0s9u1BwAAAACgPQhyAAAA9IGDTx3UowOPKjeTkyR5tqd0Ji0zbcowoimrPNuTa0XTVpXmSlGAI5B0UBKzVQEAAAD4/9m7nx8nrnz//68qbHcPbpLml2K7I1CkkGQGIUEPRLO4CBYjZTUb6EgMLCLBH5D9laIoEv9AVncHGwQzILKOxGbQjViMgqJRIvT5IqGZSB27J81wG/yjy1Vl13fhdsc4/lE/7XL38yFZuKvOqXP8rjKL8/Y5B9iBzGl3AAAAAP5c/eaqnJqj6mpV1XJVr8qvVCvXVK1UVVurqb5eV2O9oc0Xm2q8aMjzPF17dG3a3QYAAAAAIDEkOQAAAGZE6UxJV76+otrzmjb+taFquZPsqFVqnSTHv+uqr9dVrVTl1Bx98uATFZbZjAMAAAAAsHOxXBUAAMAMWfpwSZ81P9P9P9/XD3/9QZn5jLx2Zy0qwzTkWq5OXDqhC7cvTLmnAAAAAAAkjyQHAADADLp456Iu3rmote/WVP62LKkz06NwkpkbAAAAAIDdgyQHAADADCucKqhwisQGAAAAAGB3IskBAACwixiGsf3e87xQ5cZdY9h5v20DAAAAAOAXG48DAADsEt0kQzfB0Jt08Ftu3DUMw5DneduvYceHtQ0AAAAAQBDM5AAAANhFuomHcYmGUeWGnesmMkZdL4jK44rKj7f2GzldUnG5OLDc0S8DX1qS9OOn4+v++Onwc8Pqjqozql6SbQYRtY1B9f3U7a836faSbDPO+9Z7raDPWpLP5rD6SdyLuJ/RsN+fKPciSrsAAAB4HUkOAAAAxCaOJanurdzTk/tPlN2blQxJW5dxGo6Of3xcK3dXYuipP0e/HDwQOWowuHsu7gHMSbQZtY1h9YfFcVybo4zra9z9DNtmnPfNb6zCxjSKuGOaRDzjjEsc92JcbAAAAOAPSQ4AAADEZtASVf2GHV99tKqb524qfyivI2ePKLeQk2Eaajtt2TVb1oalZw+e6frcdV395qpKZ0qSxg9o+hlEHDWYOmogMmw9v/3yW8dvm0m2MexX7UHjEXQguv+aSfdzUJv91/VTPmibYcQV03HCxjRsvajxDBuXKII+pwAAAPCPPTkAAAAwMaMSHLf/dFvF5aLe+eM7WvpwSaXTJRVOFnT4t4d14NgBLb6zqIPvHVT+rbxufXRLP/39p0T7Om6QNEjiYxL9SUsb/eX8JAzCDqoHGQAfV87vckNB73tc9y1o0m4aA+dBYxqmXpR4xhUXv/cijucUAAAA45HkAAAA2EW6y0mN2/h7VDm/1xhUb9gSVjfP3dSBdw+ouFxU6XRJb//hbRV/X9Th3x3W/nf3a/Hoot5YekMLhQXtK+5T9jdZ3Th7I1D7SI7fBMCkl1Hqby+t/fQrjQPjYWPKvfhFWj8jAADArGC5KgAAgF2iu1F4NzkxbGmpUeX8nOv9u/f6vf/2nr+3ck/5w3kdfO+gDn1wSIXlghaPLqplt1T/ua7MfEaGYajtttWyW3ItV07DkfXK0leXvtKFv1yIJ0ATFtfmx0heUvusBG1/Wu3xbP6ChAQAAED6kOQAAADYRYbNpOg/PmrT8DDnRtV5cv+Jjpw9ovxbee1b2qfFo4t688ibsjYsteyWnIYju2ort5DrvPI5ZfNZze+f1/d3v08syRF1Q+w4l2DyWzdJDO7+Iq4kgJ+YTjvBkpY++JH0MzorcQAAANhtSHIAAABgaiqPK8ruzSq3kFN2b1Zm1lTLbsnasORarrz21syRPYbMjCkza8rMmcrkMp3XfEZr362pcKoQqR9hNo0eVTfoxtVxDc5OYvA1DQO8P37aidmgTZuTHOgedu2og99hNjpPyiTjmZQk4xXk2tN6TgEAAHYb9uQAAADYBd5//31VKhWtr6/r4cOH28en/b78uCwZkmEa8tqenLqj+s91VctVNZ43ZNdsuZarttuW1/Ykb2vJK1My9nTqlL8tBw9IwoImTXqPBR38nMSvy9P8C/buIHL3NQndDayjbGTtJ6aTHgiP+9mcpKSf0aiffRrPKQAAwG7BTA4AAIAd7vPPP1e5XH5tz42uNLyXJ7Wdtpyao80Xm8rMZ+Q0HBmGIddy1XzVlFN35FquWk6rk/BoeZ2kR0yGDYwO+xX2uLp+6g26DgkO/4ZtTJ30htXDEgFB7nmQBEca4h7m2ZykSSU4wlx/Ws8pAADAbkKSAwAAYBcolUrb78+fP5+a96XTnX7ZNVubG5uaW5+TYRiyq7aMPZ3Nxp26o+arpuy6LafxerLDMA2Vzvzy2eIWdPA6ar0gdnOCo1ea+zZI0JiOGgSfhfuTtEnGIMq92M33CAAAIGkkOQAAADA1xeWinIYja8NS43lDmbmM2m5buYWczIwpr+3JtVzZdVvNl03Zta1ER8OVa3VehZPR9uOYRSQ4Rktr39Par1k16/Gc9f4DAACkBUkOAAAATNXxj4/r2YNnncTGns7G47mFnMysKXlSy2nJaTiya51ER3dWh/XS0olLJ6bd/dj4XbYmjQmOcTNX0jKYG7afcSzXFCamcV0rqnF7zESJadhnZpIxSNO9AAAAwK+x8TgAAACmauXuipyGo2qlqtpaTdVyVdWfqqqWO3/X/11XY72hzf9savPFppovm7JeWlJbunD7QqJ9Gza4O27AO2i93uPTHlCN2kb/Z0xqz4FxsRzX/7D9HFQuTQPyYUV9NgddI2xM/X6/0hzPrqjPKQAAAMZjJgcAAACm7uo3V3Xro1uqrlblNBxl81llchnJlLyWp5bTkttwt2dweJ6na4+uxdb+uEHVUb80T6LeIL31xv2yPqwobfTOdhhUd1wCYNzxuGIZtp/j6g0zifs2qs1Rx/vbDPtsJhXTcfc8aDyjPGtRsLk4AABAspjJAQAAgKkrnSnpytdXVHte08a/NjqzOcpV1Sq17dkc9fW6qpWqnJqjTx58osJy8ntx/Pjp8AH9uOsNqzNLJrnp8qi2/NyfIMfjaDPtoj6bScR0J9jJzwwAAEBaMJMDAAAAqbD04ZI+a36m+3++rx/++oMy8xl5bU+SZJiGXMvViUsnfC9R5WcAMcogY9i6k6436TaCXmMa9yBK3Ul+vrDXnNYzNol2p/W8RLkmyQwAAIBkkeQAAABAqly8c1EX71zU2ndrKn9bltSZ6VE4mfzMDQAAAADAbCHJAQAAgFQqnCqocIrEBgAAAABgOJIcAAAAwA4VZMNjltQBAAAAMIvYeBwAAAAAAAAAAMwkZnIAAAAAOxSzMwAAAADsdMzkAAAAAAAAAAAAM4kkBwAAAAAAAAAAmEkkOQAAAAAAAAAAwEwiyQEAAAAAAAAAAGYSSQ4AAAAAAAAAADCTSHIAAAAAAAAAAICZRJIDAAAAAAAAAADMJJIcAAAAAAAAAABgJpHkAAAAAAAAAAAAM4kkBwAAAAAAAAAAmEkkOQAAAAAAAAAAwEwiyQEAAAAAAAAAAGYSSQ4AAAAAAAAAADCTSHIAAAAAAAAAAICZRJIDAAAAAAAAAADMJJIcAAAAAAAAAABgJpHkAAAAAAAAAAAAM4kkBwAAAAAAAAAAmEmZaXcAAIBZZxjG9nvP8yKVMwzjV+fG1fPbPgAAAAAAwE7DTA4AACLoJhi6yYXehEPQcsOOeZ63/eov038eAAAAAABgN2EmBwAAEXWTC4OSEH7LdZMV/cfHzQwJmtioPK6o/LgsSSqdLqm4XBxY7uiXgS677cdPx9f98dPx1xl0jSTr+TXss/ltI67PNen2kmozajxHXW/cNYLGNOpzPal7EfczGqatoG0n/b0FAAAAsLOR5AAAYMrGJStGLUfld6mqeyv39OT+E2X3ZiVD0lZRp+Ho+MfHtXJ3JVznQzj6ZfBB795zQeuOai+IKH0bVT9sPEYZ19e4+xmmzajxDNJ+mHJxijumw+ol8f1JUpTnFAAAAAC6SHIAADBFfmZj9J7vLz/qnCStPlrVzXM3lT+U15GzR5RbyMkwDbWdtuyaLWvD0rMHz3R97rqufnNVpTMlSeMHQv0MPo4ahB036B1X3VF1whgXFz+Dz2H61z0XdCB6WJyS6uegNvuvO65s0PbCiium44SNadh6UWMaJC5+EjRh/q+YxP0HAAAAsHOwJwcAAFNmGMb2q/t3HFYfrer2n26ruFzUO398R0sfLql0uqTCyYIO//awDhw7oMV3FnXwvYPKv5XXrY9u6ae//xRL28OETY6ErRvnAKmfgeGg1/CTMAg7qB5kAHxcOb/LDQW5d3HE02/7/deexsB52GczSL0oMZ10XOJ4TgEAAABAIskBAEBkfpMTg8r1bhreu2dHf50wbp67qQPvHlBxuajS6ZLe/sPbKv6+qMO/O6z97+7X4tFFvbH0hhYKC9pX3Kfsb7K6cfZG6Pamxe8A+zSW4+ltN239628vrf30K40D42FjOuv3Is5+pfUzAgAAAEgPlqsCACCC7mbh3WTEsOWjRpXzc/3ev/2cu7dyT/nDeR1876AOfXBIheWCFo8uqmW3VP+5rsx8RoZhqO221bJbci1XTsOR9crSV5e+0oW/XAgRDexGYfbPSKL9abWXxuRKWhAbAAAAAJPATA4AACLqn4nRe9xPuVF1xtUbdu7J/Sfa/+5+5d/Ka9/SPi0eXdSbR97U3kN7Nb84r7k35pRbyP3yyueUzWc1v39e39/9PsjHD2TUgPS4X6dPezB9FH5t/oujX77+CnuNIOWm+UxE+ZyTNAt9BAAAAIAwmMkBAMAOU3lcUXZvVrmFnLJ7szKzplp2S9aGJddy5bW3ZpXsMWRmTJlZU2bOVCaX6bzmM1r7bk2FU4VI/fCT0Bh0fNSgcRoTHL3S0L/eGI7aoD1uSSSnouzhkoRJxjMpScfL7/2e1nMKAAAAYOchyQEAQEjvv/++KpXK9t/FYlGSXjvm97hpmvp//+//6dy5c5Kkhw8fhn7/tzt/kwzJMA15bU9O3VH957padkte25Nds+VartpuW17bk7ytvT9MydjTqVP+thw5ybFbpGE2wTDTGCyOOmDtN56T/mzDNsju9mPQYH1apPkZlUhqAAAAAIiGJAcAACF8/vnnKpfLQ5eQGmTU8f4lp6K87xyQ2k5bTs3R5otNZeYzchqODMOQa7lqvmrKqTtyLVctp9VJeLS8TtIjJsMGVIf9ert7bljdUfWmJa2Dx8OW/kp6w+pRiQA/9y5ogiMNce9NdKRRmmLVb1rPKQAAAICdhSQHAAAhlUqlWI8XCr/MnDh//nz495fP65//80/ZNVubG5uaW5+TYRiyq7aMPZ3Nxp26o+arpuy6LafxerLDMA2VzgzuaxyGDXqPG4wNMlg+CWkePO5Kc9/6hYnnqEHwWbg/SZt0DMK2t5vvEQAAAIDoSHIAALDDFJeLchqOrA1LjecNZeYyartt5RZyMjOmvLYn13Jl1201XzZl17YSHQ1XrtV5FU6yVNUoszyAnsa+p7FPs27WYzrr/QcAAAAwOea0OwAAAOJ3/OPjqparaqw3VFurqVqudl6VqmprNdXX62o8b2jz/zbVfNncntVhvbR04tKJaXc/kHFL28Q9WBr0epPuX1hh+xm132E+/4+fDn/1l0naqNkkUWMa9pmZxjPF0lIAAAAApoUkBwAAO9DK3RU5DWc7qVEtV1X9qZPoqK3VVP93XY31hjb/s6nNF51Eh/XSktrShdsXEu3buMHQcQO7QerGPfAadfA46f6Nu27YTb399nNQuXH7rPjpz7T5eSYnFVO/359pxTSO5cam/RkAAAAAzBaWqwIAYIe6+s1V3frolqqrVTkNR9l8VplcRjIlr+Wp5bTkNtztGRye5+nao2uxtT9uMHbQr9e7dfz8Oj5I3TgGS3uvO6n++U34jEogBBG2n37v3bD+BY1nFGFjGjYhlVRMx93zoDGN8qxFwQwQAAAAAFExkwMAgB2qdKakK19fUe15TRv/2thesqpWqW3P5qiv11WtVOXUHH3y4BMVlpPfi2PUMkJhz3XPBzk+aZPsX9gYjqubVJtpN+65m0ZM0yTuDcd3wjMDAAAAYHKYyQEAwA629OGSPmt+pvt/vq8f/vqDMvMZeW1PkmSYhlzL1YlLJ3wvUeVn4DGOwcmw10hyYHQanytKm9OoG3RfjSSETRxEvW5S9ScV01msCwAAAAASSQ4AAHaFi3cu6uKdi1r7bk3lb8uSOjM9CieTn7kBAAAAAACQFJIcAADsIoVTBRVOkdgAAAAAAAA7A0kOAACwKwXZ8JgldQAAAAAASCc2HgcAAAAAAAAAADOJmRwAAGBXYnYGAAAAAACzj5kcAAAAAAAAAABgJpHkAAAAAAAAAAAAM4kkBwAAAAAAAAAAmEkkOQAAAAAAAAAAwEwiyQEAAAAAAAAAAGYSSQ4AAAAAAAAAADCTSHIAAAAAAAAAAICZRJIDAAAAAAAAAADMJJIcAAAAAAAAAABgJpHkAAAAAAAAAAAAM4kkBwAAAAAAAAAAmEkkOQAAAAAAAAAAwEwiyQEAAAAAAAAAAGYSSQ4AAAAAAAAAADCTSHIAAAAAAAAAAICZRJIDAAAAAAAAAADMJJIcAAAAAAAAAABgJpHkAAAAAAAAAAAAMykz7Q4AwG5jGMb2e8/zIpUzDCPQOb9tAwAAAAAAALOAmRwAMEHdJEM3wdCbdAhabljdUeU9z9t+jaoPAAAAAAAAzAJmcgDAhHUTF+MSDaPKdRMWo5IZ/efCzNyoPK6o/LgsSSqdLqm4XBxY7uiXgS8tSfrx0/F1f/x0+LlhdUfViVIval2/orYxqH6Yzzbp9pJsM8771nutoM9a2GczSv0k7kXcz2jUuIy6ziS+swAAAACA6SHJAQAzJugSVf3nu0aVu7dyT0/uP1F2b1YyJG0VdRqOjn98XCt3V0L1PYyjXwYbuOw9F2e9qHX9itrGsPrD4jiuzVHG9TXufoZtM8775jdWYWMaRdwxTSKek47LJL6zAAAAAIDpIskBADMkSoJDej2xMaj86qNV3Tx3U/lDeR05e0S5hZwM01Dbacuu2bI2LD178EzX567r6jdXVTpTkjR+QNPPIOKowdRRg7Rh6o3rb9D2/Nb1K2wbw2YY+O1b91zQgej+aybdz0Ft9l/XT/mgbYYRV0zHCRvTsPWixjNoXPwkafy0N6heUvceAAAAADAZ7MkBADPGMIztV/dvP+fGWX20qtt/uq3iclHv/PEdLX24pNLpkgonCzr828M6cOyAFt9Z1MH3Dir/Vl63Prqln/7+U7wfrs+4QcugiYgo56LW9SuONvrL+flcYQfVgwyAjyvnd8mioPc9rvsWNGk3jYHzoDENUy/qd2iScQn7/wQAAAAAYHaQ5ACACfObgBhUrnfj8N49O8ad89PezXM3deDdAyouF1U6XdLbf3hbxd8Xdfh3h7X/3f1aPLqoN5be0EJhQfuK+5T9TVY3zt4I+OmRFL8JgGkvF5TWfvqVxoHxsDGd9XshpbtvAAAAAIDJYLkqAJig7obg3YTDsOWjRpWL2nbv3133Vu4pfzivg+8d1KEPDqmwXNDi0UW17JbqP9eVmc/IMAy13bZadkuu5cppOLJeWfrq0le68JcLkfsHjDLt/ROmnRxKY3IlTYgPAAAAAOxeJDkAYMKGJSz6j/tJbIwqM+jcsPJP7j/RkbNHlH8rr31L+7R4dFFvHnlT1oallt2S03BkV23lFnKdVz6nbD6r+f3z+v7u94klOaJuiB1lX4OwbSaJX63/Iq4kgJ+YTjvBkpY++DFLz+isxBQAAAAAMBpJDgDY5SqPK8ruzSq3kFN2b1Zm1lTLbsnasORarrz21qySPYbMjCkza8rMmcrkMp3XfEZr362pcKoQqR9hNo0eVTfKwOW06qapDT99OPrl4E2bkxzoHnbtqAPWUfZjidsk45mUScQr6D2P+/8JAAAAAEA6sCcHAExIpVJ57RX1+D/+8Y/tYw8fPgz9/m93/iYZkmEa8tqenLqj+s91VctVNZ43ZNdsuZarttuW1/Ykb2t/D1My9nTqlL8tRw9QzIIODEcZJJ/EL8LT/KvzbrKj+5qE7obSUTay9hPTSScYxm3knuaER5qf0WHSHE8AAAAAgD/M5ACACfjiiy907NgxnT9//lfn/C5f1X+893yU950DUttpy6k52nyxqcx8Rk7DkWEYci1XzVdNOXVHruWq5bQ6CY+W10l6xGTYwOiw2QLj6vqp11t2VB+SqpumNsIYtjF10htWD0sExH3P0xT37udLqzTFapio/08AAAAAANKJJAcATMjTp091+fLlXx0vlUoDy4873nu+N3kS+P3l8/rn//xTds3W5sam5tbnZBiG7KotY09ns3Gn7qj5qim7bstpvJ7sMExDpTOD+xqHoIPXQeuR4IguzX0bJI5ljsJeayeaRgziajPs/y8AAAAAgPQgyQEAu1xxuSin4cjasNR43lBmLqO221ZuISczY8pre3ItV3bdVvNlU3ZtK9HRcOVanVfhZLT9OKaFBEdy0tr3tPZrVhFPAAAAAMC0sScHAEDHPz7e2YNjvaHaWk3VcrXzqlRVW6upvl5X43lDm/+3qebL5vasDuulpROXTky7+6HstATHuCWi0jIYHbafcfQ7TEyHvfrLJG3UbJKoMQ37zEzrmUrzsl0AAAAAgMkjyQEA0MrdFTkNZzupUS1XVf2pk+iordVU/3ddjfWGNv+zqc0XnUSH9dKS2tKF2xcS7du4Adig9XrP7ZQEx7D6w/6OS9SESth+DiqX1gH5IPw865OKqd/v1zTjGWTvlbDnAQAAAADpx3JVAABJ0tVvrurWR7dUXa3KaTjK5rPK5DKSKXktTy2nJbfhbs/g8DxP1x5di639cYONwzYNHqe/Xm8dP7+Oj6uuX1Ha6N2celDdcQmAccfD3oNB/QjTz3H1hpnEfRvV5qjjo57PIJKKadzfgyjPWlhh/p8AAAAAAMwOZnIAACRJpTMlXfn6imrPa9r418b2klW1Sm17Nkd9va5qpSqn5uiTB5+osJz8XhzDlgManDlB3gAAIABJREFUNyg5qWWE0mbUYPYk2/Jzf4Icj6PNtBvV/2nFNG3CLuM26nwaPycAAAAAwD9mcgAAti19uKTPmp/p/p/v64e//qDMfEZe25MkGaYh13J14tIJ30tU+Rk8jDLAGKbupNubRhtBrzGtmIStO8nPF/aak/ps02h3mt+hSX/nAQAAAADpR5IDAPArF+9c1MU7F7X23ZrK35YldWZ6FE4mP3MDAAAAAAAA8IskBwBgqMKpggqnSGwAAAAAAAAgnUhyAACQoCAbSbOsDgAAAAAAQDBsPA4AAAAAAAAAAGYSMzkAAEgQszMAAAAAAACSw0wOAAAAAAAAAAAwk0hyAAAAAAAAAACAmUSSAwAAAAAAAAAAzCSSHAAAAAAAAAAAYCaR5AAAAAAAAAAAADOJJAcAAAAAAAAAAJhJJDkAAAAAAAAAAMBMIskBAAAAAAAAAABmEkkOAAAAAAAAAAAwk0hyAAAAAAAAAACAmUSSAwAAAAAAAAAAzCSSHAAAAAAAAAAAYCaR5AAAAAAAAAAAADOJJAcAAAAAAAAAAJhJJDkAAAAAAAAAAMBMIskBAAAAAAAAAABmEkkOAAAAAAAAAAAwk0hyAAAAAAAAAACAmZSZdgcAAAAwmGEY2+89zwtVLuy53jKj2gYAAAAAYJqYyQEAAJBC3QREN8HQm5DwW66boOi+/J7rvzYAAAAAAGnFTA4AAICU6iYuhiUhxpUbNQNj3OyMbhLEb6Kj8rii8uOyJKl0uqTicnFguaNf+rrcr/z46fi6P346/NywuqPqjKoXpU2/bfsR9nONqu+nbn+9SbeXZJtRYzrsWkGftSSfzWH1k7gXcT+jQe7DNO89AAAAJockBwAAwA4WZrmqIEtU3Vu5pyf3nyi7NysZkraqOQ1Hxz8+rpW7K+E7H9DRLwcPRo4aDO6em8VBzKifa1j9YXEc1+Yo4/oadz/Dthnns+I3VmFjGkXcMU0inlHiMu17DwAAgMkiyQEAALCDjUpeDDrnN8Gx+mhVN8/dVP5QXkfOHlFuISfDNNR22rJrtqwNS88ePNP1ueu6+s1Vlc6UJI0f0PQziDhqgHLUIG3Yen77lUTdsNce97mGzTAIGo+gA9H910y6n4Pa7L+un/JB2wwjrpiOEzamYetFjWfYuAxqO+m+AgAAYDrYkwMAAACvMQxj+9X9u9fqo1Xd/tNtFZeLeueP72jpwyWVTpdUOFnQ4d8e1oFjB7T4zqIOvndQ+bfyuvXRLf30958S7fO4QdIgiY9ZEUdSpr+cn4RB2EH1IIPK48r5XYop6H2PK9EVNGk3jecwaEzD1IsSz7BxCfu8JZXkBAAAQPJIcgAAAKTUsCSD33Kj6g0717sZee9eH71unrupA+8eUHG5qNLpkt7+w9sq/r6ow787rP3v7tfi0UW9sfSGFgoL2lfcp+xvsrpx9sboD4uJ8ZsAmPQySv3tpbWffqVxYDxsTGf9XgwyS30FAADAaCxXBQAAkELdpaO6yYhhy06NKjdqI/Igm5T3urdyT/nDeR1876AOfXBIheWCFo8uqmW3VP+5rsx8RoZhqO221bJbci1XTsOR9crSV5e+0oW/XAgRjemLc/PjNA5+7yTT3j9h2skhni8AAADsNiQ5AAAAUmpY4qH/+KgERdhzw8o8uf9ER84eUf6tvPYt7dPi0UW9eeRNWRuWWnZLTsORXbWVW8h1Xvmcsvms5vfP6/u73yeW5Ii6IXacSzAlWTdoG4gvCeAnptNOsKSlD37M0jM6S30FAADYjUhyAAAAwJfK44qye7PKLeSU3ZuVmTXVsluyNiy5liuvvTWrZI8hM2PKzJoyc6YyuUznNZ/R2ndrKpwqROpHmE2jR9UNunF1kAHPKHXjkIaB7h8/7XzuQZs2JxmPYdeOmgQIs9F5Uqb9fMUh7ngl+byl4fsEAACAX2NPDgAAgJSpVCqvvYIc/8c//rF9/OHDh7G+Lz8uS4ZkmIa8tien7qj+c13VclWN5w3ZNVuu5arttuW1Pcnb2vvDlIw9nTrlb8tRwxO7oEmT3mNJ1Y0izb/k7w4+d1+T0N3AOsoG335imoYE1iSerzhM6hmN43lL8/cJAAAAHczkAAAASJEvvvhCx44d0/nz5391zs/yVUm/lye1nbacmqPNF5vKzGfkNBwZhiHXctV81ZRTd+RarlpOq5PwaHmdpEdMhg02Dvv19ri6fuoNuk7YQdModcdJ64DssI2pk96welgiIMg9D5LgSEPck3y+4jCJWMX1vKXpvgIAAGA4khwAAAAp8/TpU12+fPlXx0ul0sDyvcd73/cmSuJ4XzrdubZds7W5sam59TkZhiG7asvY09ls3Kk7ar5qyq7bchqvJzsM01DpzODPEIegg9dR66XNLAzIprlvgwSN6ajB81m4P0mbdAyitMP9AgAAmB0kOQAAAOBLcbkop+HI2rDUeN5QZi6jtttWbiEnM2PKa3tyLVd23VbzZVN2bSvR0XDlWp1X4WS0/Tgw2CwPyKa172nt16xKSzxnbWYOAAAAxmNPDgAAAPh2/OPjnT041huqrdVULVc7r0pVtbWa6ut1NZ43tPl/m2q+bG7P6rBeWjpx6cS0ux+bKMsBxb2UUNAB2XFL9qRlgDdsP+Pod5iYDnv1l0man31iwsY07DOTlmfKj1nqKwAAADpIcgAAAMC3lbsrchrOdlKjWq6q+lMn0VFbq6n+77oa6w1t/mdTmy86iQ7rpSW1pQu3LyTat3EDsHHV6z0+blA3TN0gog7I9vcz6b1Chh0f1/+w/RxUbicMyMfxfMUVU7/fr0nGczckYwAAAPALlqsCAABAIFe/uapbH91SdbUqp+Eom88qk8tIpuS1PLWcltyGuz2Dw/M8XXt0Lbb2xw2qDttcfJyw9YZJevPn3uv7+fV+/7FunUF1oyRvhtUPE4+w/RxXb5goMQ0rbEzj2Pg+zpiOu+dB4xnlWRvX3rjyk7r3AAAAiAczOQAAABBI6UxJV76+otrzmjb+tbG9ZFWtUtuezVFfr6taqcqpOfrkwScqLCe/F8ew5YDGDUqGqTdu6aEodSdl1GD2JNvyc3+CHI+jzbSL+nwlEdM02cn3HgAAAL/GTA4AAAAEtvThkj5rfqb7f76vH/76gzLzGXltT5JkmIZcy9WJSyd8L1HlZ+AxyuBk2LrTaHOS1w96jWnFY1L3b5IJnqhtRu3rJNqd5vcnDfceAAAAk0GSAwAAAKFdvHNRF+9c1Np3ayp/W5bUmelROJn8zA0AAAAAAEhyAAAAILLCqYIKp0hsAAAAAAAmiyQHAACYaYZhbL/3PC9wOT/1o9QF/AqyUTJL6wAAAABABxuPAwCAmdVNMnQTDL1JB7/lPM8bmxzplulPcPQeH9Y2AAAAAABIDjM5AADATOsmHsYlGvyW69VNZIy6XhCVxxWVH2/tW3G6pOJyMfA1sHMxOwMAAAAAgiPJAQAAMMKoJan8Lld1b+Wentx/ouzerGRI2irqNBwd//i4Vu6uxNpnAAAAAAB2C5IcAAAAIwxaosrPOUlafbSqm+duKn8oryNnjyi3kJNhGmo7bdk1W9aGpWcPnun63HVd/eaqSmdKyX8gAAAAAAB2EJIcAAAACVh9tKrbf7qt4nJRB987qPxbeWX3ZuW1PTk1R5sbm2o8byi3kFO1UtWtj27pytdXpt1tAAAAAABmCkkOAAAw07ozKMbts+G3XNB2h7l57qaKy0UVl4s69MEh7VvaJzNryqk72nyxqbn1OWXmMjL3mJKk6mpVN87ekP47lu4BAAAAALArkOQAAAAzq5u06CYuhi0fNa5c//thm5T3L1U17Ny9lXvKH87r4HsHdeiDQyosF7R4dFEtu6X6z3Vl5jMyDENtt62W3ZJruXIajqxXlux7tsQWHQAAAAAA+EKSAwAAzLRhsyn6j/stF+T8sHNP7j/RkbNHlH8rr31L+7R4dFFvHnlT1oallt2S03BkV23lFnKdVz6nbD6r+f3zsp/YI/sDAAAAAAB+YU67AwAAADtJ5XFF2b1Z5RZyyu7NysyaatktWRuWXMuV196aVbLHkJkxZWZNmTlTmVxGmVxGykjGWjxLagEAAAAAsNOR5AAAADOpUqm89uo9/vTp0+2/Hz58ONH35cdlyZAM0+hsMl53VP+5rmq5qsbzhuyaLddy1Xbb8tqe5G0tk2Wqk/gwTKkcKiQAAAAAAOw6LFcFAABmzhdffKFjx47p/Pnzvzrned6v9s6Y+HtPajttObXOJuOZ+YychiPDMORarpqvmnLqjlzLVctpdRIeLa+T9AAAAAAAAL6R5AAAADPp6dOnunz58q+Ol0ql1/7uTYRM4n3ljc6sErtma3NjU3PrczIMQ3bVlrGns9m4U3fUfNWUXbflNPqSHZ4nr0SyAwAAAAAAP0hyAAAAxKi4XJTTcGRtWGo8bygzl1HbbSu3kJOZMeW1PbmWK7tuq/myKbu2lehouJ09O1xPKkz7UwAAAAAAMBtIcgAAAMTs+MfH9ezBs05iY09n4/HcQk5m1pQ8qeW05DQc2bVOoqM7q8N6aUnHp917AAAAAABmB0kOAACAmK3cXdH1ueuqVqqSJNdylcvnZOZMGUZnySrXcuU0OstWNV82OwmOtqQVSaxWBQAAAACAL+a0OwAAALATXf3mqpyao+pqVdVyVa/Kr1Qr11StVFVbq6m+XldjvaHNF5tqvGjI8zxde3Rt2t0GAAAAAGCmkOQAAABIQOlMSVe+vqLa85o2/rWharmT7KhVap0kx7/rqq/XVa1U5dQcffLgExWW2YwDAAAAAIAgWK4KAAAgIUsfLumz5me6/+f7+uGvPygzn5HX7qxFZZiGXMvViUsndOH2hSn3FAAAAACA2USSAwAAIGEX71zUxTsXtfbdmsrfliV1ZnoUTjJzAwAAAACAKEhyAAAATEjhVEGFUyQ2AAAAAACIC0kOAACAlDIMY/u953mBy/UeH3UdwzBG1hvVNgAAAAAA08TG4wAAACnUTTR0EwyDEhbjynme99prVP1+4+oBAAAAAJAGzOQAAABIqW6CwfO8ockIv+X6Z2v0Hht1bb8qjysqP97ab+R0ScXl4sByR78Md/0fPx1f98dPh58bVndUnXH1/daNo36Q6wa5dth+9debdHtJthk1psOuNa5+0JhG+T4Mq5/EvYj7GZ3E9y6p7ysAAACSQZIDAABgFxqU9Og/3zWq3L2Ve3py/4mye7OSIWmrqNNwdPzj41q5uxJXl8c6+uXggchRg8Hdc8MGMKPU9VM/ysBpUn0b1a+wSaoocQjTz7BtRo2p32uFKRenuGM66e/eOGHbjfP+AwAAYHJIcgAAAOxwg/bcGJW4GDbjo9fqo1XdPHdT+UN5HTl7RLmFnAzTUNtpy67ZsjYsPXvwTNfnruvqN1dVOlOSNH5A088A4qjByVGDtEHrjfsVvp82/dSPYlw8/Qw+9573+5m654J+hmExTqqfg9rsv66f8kHbDCOumI4TNqZh60WNZ9C4hP3exvl9BwAAwGSxJwcAAMAuZBjG9qv7t1+rj1Z1+0+3VVwu6p0/vqOlD5dUOl1S4WRBh397WAeOHdDiO4s6+N5B5d/K69ZHt/TT339K6qNIGj9IGiTx4beM3+WAorQ9ip+B4aDX8JMwCDuoHmQAfFy5pGIfR0z9tN9/3WkMmgeNaZh6UeIZNS5hv3dJfV8BAACQHJIcAAAAKeU3ARE0UTFoQ/IgG4zfPHdTB949oOJyUaXTJb39h7dV/H1Rh393WPvf3a/Fo4t6Y+kNLRQWtK+4T9nfZHXj7A3f10ey/CYAJr2MUn97ae2nX2kcFA8b01m/FwAAANjZWK4KAAAghbobgncTF8OWmxpVrr+sX/3Jkt7691buKX84r4PvHdShDw6psFzQ4tFFteyW6j/XlZnPyDAMtd22WnZLruXKaTiyXln66tJXuvCXC4H6MqtYv396ph37aSeHeOYAAACw2zCTAwAAIKX6Z1v0HvdTblDZYe0Mu17/uSf3n2j/u/uVfyuvfUv7tHh0UW8eeVN7D+3V/OK85t6YU24h98srn1M2n9X8/nl9f/d7vx89sKgbYo9aSmnUZsvD6g4q2/tKGr+o/0VcsfdTd9oJlm4fZuH+J9XHsN/bOL/vAAAAmCxmcgAAAMCXyuOKsnuzyi3klN2blZk11bJbsjYsuZYrr701q2SPITNjysyaMnOmMrlM5zWf0dp3ayqcKkTqR5hNo0fVHbdvwKhB4zDt9R5PesA0DQOyvTEctbl73JKKfdg9HZIwyXgmJalN3MN8b6N+3wEAADAdJDkAAABS5v3331elUtn+u1gsStJrxyZx/NmzZ/qv//ovSdLDhw+18P8tSIZkmIa8tien7qj+c10tuyWv7cmu2XItV223La/tSd7W0lemZOzp1Cl/W46c5IjboMH3uE1jcD+NA7LTGISPI/Z+YjrpzzZs9lG3H5N4rsNK8zMKAACA2UOSAwAAIEU+//xzlcvlQEtPJXW89+/t957Udtpyao42X2wqM5+R03BkGIZcy1XzVVNO3ZFruWo5rU7Co+V1kh4xGTYwOmy2wLi6o+qNGoz1096wuqNmN0SR1sHjYUsBJb1hdRyxD5LgSEPcexMdaTSJWIX93sbxfQcAAMDkkeQAAABImVKplIrjvX+fP39elTc6Mz3smq3NjU3Nrc/JMAzZVVvGns5m407dUfNVU3bdltN4PdlhmIZKZwa3HYewiYNh9cYNxiaVqAgrTQPtw6S5b4MEjemo5MIs3J+kTTvB0T2+E77vAAAA+AVJDgAAAPhSXC7KaTiyNiw1njeUmcuo7baVW8jJzJjy2p5cy5Vdt9V82ZRd20p0NFy5VudVOJmupap2ilkeQE9r39Par1lFPAEAAJAUc9odAAAAwOw4/vFxVctVNdYbqq3VVC1XO69KVbW1murrdTWeN7T5f5tqvmxuz+qwXlo6cenEtLs/MZMcyA06eDxuiai0DEaH7Wcc/Q4T02Gv/jJJGzWbJGpMwz4zaXmmAAAAsDOR5AAAAIBvK3dX5DSc7aRGtVxV9adOoqO2VlP933U11hva/M+mNl90Eh3WS0tqSxduX0i0b+MGYIPWi3rdUeXiGvSNep3+viW1l0PUhErYfoaJ/SwMyPt5JicVU7/fn0nHM+7/D9K8zwkAAMBux3JVAAAACOTqN1d166Nbqq5W5TQcZfNZZXIZyZS8lqeW05LbcLdncHiep2uPrsXW/rjBxlEbCgep17uBs59fxw867qd+WL3XDNq/cX0blwAYdzzsPRjUjzD9DBv7KDENK2xMwz5TScV03D0PGs+wcQn7vY36fQcAAMD0kOQAAABAIKUzJV35+opunL0h65Wl+f3zyuQyMvYY8tqe2m5bruVuz+C49uiaCsvJ78XhZ3A2SL3ec0EGg/20nYaB0kn2LUpbYeumOfZRjXqmiWn4723U7zsAAACmgyQHAAAAAlv6cEmfNT/T/T/f1w9//UGZ+Yy8tidJMkxDruXqxKUTvpeo8jswG1bUAcpptp3kdYNeY1pxCFt3kp8v7DUn9dmm0e60vzfTii0AAAAmiyQHAAAAQrt456Iu3rmote/WVP62LKkz06NwMvmZGwAAAAAAkOQAAABAZIVTBRVOkdgAAAAAAEwWSQ4AAHYBwzC233ueF7jcqPq95/yUGdU+sJsF2Uia5XQAAAAAoMOcdgcAAECyugmGbnJhUFJiXDnP84YmJ7rnhpUxDGPkeQAAAAAAgLCYyQEAwC7QTS54njc0yRGk3DDdhMawv/2oPK6o/Hhrb4fTJRWXi4H7AcwiZmcAAAAAQHAkOQAAQKL8LlV1b+Wentx/ouzerGRI2irqNBwd//i4Vu6uJNxTAAAAAAAwa0hyAACAWAybtTFuZsfqo1XdPHdT+UN5HTl7RLmFnAzTUNtpy67ZsjYsPXvwTNfnruvqN1dVOlNK/LMAAAAAAIDZQJIDAABMzeqjVd3+020Vl4s6+N5B5d/KK7s3K6/tyak52tzYVON5Q7mFnKqVqm59dEtXvr4y7W4DAAAAAICUIMkBAMAu0J1BMW6fDb/l4nLz3E0Vl4sqLhd16IND2re0T2bWlFN3tPliU3Prc8rMZWTuMSVJ1dWqbpy9If33RLoHAAAAAABSjiQHAAA7XDdp0U1cDFs+aly5/vd+NhjvT5j0lrm3ck/5w3kdfO+gDn1wSIXlghaPLqplt1T/ua7MfEaGYajtttWyW3ItV07DkfXKkn3PltiiAwAAAACAXY8kBwAAu8CwDb/7j/stF+T8sHNP7j/RkbNHlH8rr31L+7R4dFFvHnlT1oallt2S03BkV23lFnKdVz6nbD6r+f3zsp/YI/sDAAAAAAB2B3PaHQAAALtP5XFF2b1Z5RZyyu7NysyaatktWRuWXMuV196aVbLHkJkxZWZNmTlTmVxGmVxGykjG2mSW1AIAAAAAAOlFkgMAgB2sUqm89uo//r//+7/bxx4+fDix93+78zfJkAzT6GwyXndU/7muarmqxvOG7Jot13LVdtvy2p7kbS2TZaqT+DBMqRwmIgAAAAAAYCdhuSoAAHaoL774QseOHdP58+d/da53H47+Y5N43zkgtZ22nFpnk/HMfEZOw5FhGHItV81XTTl1R67lquW0OgmPltdJegAAAAAAAIgkBwAAO9rTp091+fLlXx0vlUqv/SvptWRI4u8vn9c//+efsmu2Njc2Nbc+J8MwZFdtGXs6m407dUfNV03ZdVtOoy/Z4XnySiQ7AAAAAADY7UhyAACAiSsuF+U0HFkblhrPG8rMZdR228ot5GRmTHltT67lyq7bar5syq5tJToabmfPDteTCtP+FAAAAAAAYNpIcgAAgKk4/vFxPXvwrJPY2NPZeDy3kJOZNSVPajktOQ1Hdq2T6OjO6rBeWtLxafceAAAAAACkAUkOAAAwFSt3V3R97rqqlaokybVc5fI5mTlThtFZssq1XDmNzrJVzZfNToKjLWlFEqtVAQAAAACw65nT7gAAANi9rn5zVU7NUXW1qmq5qlflV6qVa6pWqqqt1VRfr6ux3tDmi001XjTkeZ6uPbo27W4DAAAAAICUIMkBAACmpnSmpCtfX1HteU0b/9pQtdxJdtQqtU6S49911dfrqlaqcmqOPnnwiQrLbMYBAAAAAAA6WK4KAABM1dKHS/qs+Znu//m+fvjrD8rMZ+S1O2tRGaYh13J14tIJXbh9Yco9BQAAAAAAaUOSAwAApMLFOxd18c5FrX23pvK3ZUmdmR6Fk8zcAAAAAAAAg5HkAAAAqVI4VVDhFIkNAAAAAAAwHkkOAACAlDIMY/u953mhyw0633usv96ocwAAAAAApAlJDgAAgBTqJho8z5NhGDIMY2QCY1i5YfW6dYYhsQEAAAAAmAUkOQAAAFKqm2joJjCClhuV4Ihb5XFF5cdbe6mcLqm4XBxY7uiX4a7/46fj6/746fBzw+qOquPnGn7qh63nR9TPFddnmnR7SbYZx7My6Frj6geNaZTvw7D6SdyLuJ/RSXxnh10jru8tAAAA4kWSAwAAYAcbtZRV2HO97q3c05P7T5Tdm5UMSVtFnYaj4x8f18rdlQi9D+bol4MHIUcNBnfPhRmkHdVmHO2Ok9TnCvuZRgkbw1F1w9YbVTfOe+Y3VmFjGkXcMU0inlHiEtd9nMa9AQAAQHAkOQAAAHawYUtXDUpq+DnXtfpoVTfP3VT+UF5Hzh5RbiEnwzTUdtqya7asDUvPHjzT9bnruvrNVZXOlCSNH9D0M/A4ajB11CBt2Hp+6o86HrXdoH3yc/1h/fPbr+65oIPA/ddMup+D2uy/rp/yQdsMI66YjhM2pmHrRY1n0LhM6rsHAACA9DCn3QEAAADMltVHq7r9p9sqLhf1zh/f0dKHSyqdLqlwsqDDvz2sA8cOaPGdRR1876Dyb+V166Nb+unvPyXap3GDpEESH73GJV/CDLD7qeeHn4HhoNfw83nCDqoHGQAfVy6p+xZHTP2033/daQy2B41pmHpR4hk1LlG/e3HMtgIAAMBkkOQAAABIqe6SUaP24whSLi43z93UgXcPqLhcVOl0SW//4W0Vf1/U4d8d1v5392vx6KLeWHpDC4UF7SvuU/Y3Wd04e2MifcN4fhMAk16qp7+9tPbTrzQOjoeN6azfi7DSeA8BAADwayxXBQAAkELdTcS7iYtRy04NK9e/EXn/Nfrb83Pu3so95Q/ndfC9gzr0wSEVlgtaPLqolt1S/ee6MvMZGYahtttWy27JtVw5DUfWK0tfXfpKF/5yIXRMJo1fcs+mad+3aSeHeF6j22nJGgAAgJ2OmRwAAAAp5Xne9qv/uJ9y464R5tyT+0+0/939yr+V176lfVo8uqg3j7ypvYf2an5xXnNvzCm3kPvllc8pm89qfv+8vr/7fZRwjBR1Q2y/mxD3vkYZ98v2pAfiGaT9RZD7Nu46fstMM9EQ9XNOSlJ9jPrdS8M9BAAAQDDM5AAAAIAvlccVZfdmlVvIKbs3KzNrqmW3ZG1Yci1XXntrVskeQ2bGlJk1ZeZMZXKZzms+o7Xv1lQ4VYjUjzCbRo+qG6ZO7/FRS/iMGnCexCBqGgZqe+MwbOPxJCSVYAq7D0sSJhnPpCS1iXuU714avjcAAADwjyQHAABAylQqldf+LhaLEzn+8OFDnTt3buj78uOyZEiGachre3Lqjuo/19WyW/LanuyaLddy1Xbb8tqe5G0tfWVKxp5OnfK35chJjrgNGnwfZFYGlNP8S/RpxCyO++YnppP+bMM2cu/2w+9zPQ1pfUbT+p0GAADAaCQ5AAAAUuSLL77QsWPHdP78+V+dG7QcVVLHh72XJ7Wdtpyao80Xm8rMZ+Q0HBmGIddy1XzVlFN35FquWk6rk/BoeZ2kR0xGLTMzbmB30Lmw9UbNUOheN0qbYaR18HjYEkJJb1iDSwJAAAAgAElEQVQd5r71C5LgSEPcexMdaTSJWIX57qXpHgIAACAYkhwAAAAp8/TpU12+fPlXx0ul0sDycR3vTawMel863aln12xtbmxqbn1OhmHIrtoy9nQ2G3fqjpqvmrLrtpzG68kOwzRUOjO47TgEHbyOWm+UIEtZTarNNEhz3wYJGtNRyYVZuD9Jm3aCo3vcT3IyzLUBAAAwHSQ5AAAA4EtxuSin4cjasNR43lBmLqO221ZuISczY8pre3ItV3bdVvNlU3ZtK9HRcOVanVfhZLqWqtopZnnwNa19T2u/ZhXxBAAAQFJIcgAAAMC34x8f17MHzzqJjT2djcdzCzmZWVPypJbTktNwZNc6iY7urA7rpaUTl05Mu/uBpH3Zn66gg8dRltmapLD9jOO+hYlpXNeKatRnjxrTsM9MWp6pUdJ0DwEAABCMOe0OAAAAYHas3F2R03BUrVRVW6upWq6q+lNV1XLn7/q/62qsN7T5n01tvthU82VT1ktLaksXbl9ItG/DBnfHDXj7GRAfVCbKZtRxJU+iDr729yOppM64OIzrf9h+hrlvszCg7ee5mlRM/X6/Jh3PpL97AAAASA9mcgAAACCQq99c1a2Pbqm6WpXTcJTNZ5XJZSRT8lqeWk5LbsPdnsHheZ6uPboWW/vjBimDrrM/ql7vrIAgg6N+60UZ+O29btA2xvVvXAJg3PGw92BQP8L0M+x9ixLTsMLGNOxgfVIxHXfPg8YzbFwm8d0DAABAujCTAwAAAIGUzpR05esrqj2vaeNfG53ZHOWqapXa9myO+npd1UpVTs3RJw8+UWE5+b04fvx0+IB+mHrj6vupF7buJIzq2yTb8nN/ghyPo820C/vcjaufVL1JS/t3DwAAAPFiJgcAAAACW/pwSZ81P9P9P9/XD3/9QZn5jLy2J0kyTEOu5erEpRO+l6jyM+gYZWAy6qDmNNtO8rpBrzGtOIStO8nPF/aak/ps02h32t+buO4nSREAAIB0I8kBAACA0C7euaiLdy5q7bs1lb8tS+rM9CicTH7mBgAAAAAAJDkAAAAQWeFUQYVTJDYAAAAAAJNFkgMAAMw0wzC233ueF7jcuPp+6o1rG/Aj6MbmAAAAAAA2HgcAADOsm2joJhj6Ew9+ynmeNzRBYRjG9nnP8wYmNkbVBwAAAAAAyWImBwAAmGndBMOgJESYckmqPK6o/Hhr34rTJRWXi1PpB9KJ2RkAAAAAEBxJDgAAgCH6EyL9Mzb8LpV1b+Wentx/ouzerGRI2irqNBwd//i4Vu6uxNpvAAAAAAB2C5IcAAAAQ3SXqxr096CER/+x1UerunnupvKH8jpy9ohyCzkZpqG205Zds2VtWHr24Jmuz13X1W+uqnSmlPyHAgAAAABgByHJAQAAkIDVR6u6/afbKi4XdfC9g8q/lVd2b1Ze25NTc7S5sanG84ZyCzlVK1Xd+uiWrnx9ZdrdBgAAAABgppDkAAAAM607g2LcPht+y8Xl5rmbKi4XVVwu6tAHh7RvaZ/MrCmn7mjzxabm1ueUmcvI3GNKkqqrVd04e0P674l0DwAAAACAHYEkBwAAmFndpEU3cTFqaalR5frfD9ukfFi9/nP3Vu4pfzivg+8d1KEPDqmwXNDi0UW17JbqP9eVmc/IMAy13bZadkuu5cppOLJeWbLv2RJbdAAAAAAA4AtJDgAAMNOGbfjdf9xvubDX7/Xk/hMdOXtE+bfy2re0T4tHF/XmkTdlbVhq2S05DUd21VZuIdd55XPK5rOa3z8v+4k9sj8AAAAAAOAX5rQ7AAAAsJNUHleU3ZtVbiGn7N6szKyplt2StWHJtVx57a1ZJXsMmRlTZtaUmTOVyWWUyWWkjGSsTWZJLQAAAAAAZh1JDgAAMJPef/99VSqV7VdX9++nT59uH3v48OHE3v/tzt8kQzJMo7PJeN1R/ee6quWqGs8bsmu2XMtV223La3uSt7X0lalO4sMwpXKYiAAAAAAAsPuwXBUAAJg5n3/+ucrl8sAlo3r30+g/Non3nQNS22nLqXU2Gc/MZ+Q0HBmGIddy1XzVlFN35FquWk6rk/BoeZ2kBwAAAAAA8I0kBwAAmEmlUsn38fPnz0/u/eXz+uf//FN2zdbmxqbm1udkGIbsqi1jT2ezcafuqPmqKbtuy2n0JTs8T16JZAcAAAAAAH6Q5AAAAIhRcbkop+HI2rDUeN5QZi6jtttWbiEnM2PKa3tyLVd23VbzZVN2bSvR0XA7e3a4nvT/s3f/wXGc953nP92YGYAckAQJUhwMaNC0Sco2zUSkSVqOQ5PrONHdJr6tkNRGllJRitpcflQurOQu92N3FRWrtNn11tXmVIk32dglVioMFYtLXW1dUmeHsWw5spwo4soX6ZizHNqiQgIQQVIA53d3z/T90RhwBM2Pnp6emR7g/arq4qC7n36efrpHcb7feZ4n1e+7AAAAAABgMJDkAAAACNmeB/foysUrXmJjyFt4PDGakBk3JVcq22XZeVtW1kt0VEd1FBeK0p5+tx4AAAAAgMFBkgMAACBkJ549oSeHn1RmJiNJcoqOEsmEzIQpw/CmrHKKjuy8N21VaaHkJTgqkk5IYrYqAAAAAAB8MfvdAAAAgJXo5IsnZWdtZa5llJnO6M70HWWns8rMZJSdzSo3l1N+Lq/C7YLyt/NyXVePvfRYv5sNAAAAAMBAIckBAADQBemDaT3ylUeUvZnV/Jvzykx7yY7sTNZLcrydU24up8xMRnbW1qMXH1VqP4txAAAAAADQDqarAgAA6JLJQ5N6vPS4Lnzugl7/8uuKjcTkVry5qAzTkFN0tPehvTp27lifWwoAAAAAwGAiyQEAANBlx585ruPPHNfsq7OafmVakjfSI3UfIzcAAAAAAOgESQ4AAIAeSe1LKbWPxAYAAAAAAGEhyQEAABBRhmEsfXZdN9B5jY7V7m91vFndAAAAAAD0EwuPAwAARFA1yVBNMNRLSrQ6zzAMua67tC1PXNRuy6/ZqBwAAAAAAFHCSA4AAICIqiYfWiUaGp3ndwRGNanRbrlaM5dmNH1pcb2RA2lN7J+oe972p9q+tCTp6qnWZa+eanysUdlmZfxcw2/5Tsq2e912rh20XcvL9bq+btYZxrtS71qtyrfbp518HxqV78azCPsd7eZ3NsxnDwAAgN4hyQEAAID38JssOX/ivC5fuKz42rhkSFo81c7b2vPgHp149kSXW3rX9qfqByObBYOrx4IEPpvVGUbZVrp1X83aFTRJ1Y0+7Ebfd9qnfq8V5Lwwhd2n3ejPTvolSL1hPnsAAAD0FkkOAACAVWD5aI1W+5evz7H8nGsvXdOZI2eU3JzU1OEpJUYTMkxDFbsiK2upOF/UlYtX9OTwkzr54kmlD6YltQ5o+gkiNgumNgvSBi3np3wrnZRt97q11/YTfK497rc/qsfavYdGgeVutbNencuv6+f8dusMIqw+bSVonwYt12l/ttsvrUbPNKu3X88eAAAAnWFNDgAAgBWuUSIjqGsvXdO5z57TxP4J7fjMDk0emlT6QFqp+1La8uEt2rRrk8Z2jGl897iSW5M6+8BZXX/5emj119MqSNpO4qNWq+SLn1+9B627FT+B4Xav4SdhEDSo3k4AvNV53XpuYfSpn/qXX7cfgfN2+zRIuU76s9N+6dezBwAAQO+R5AAAAIio6pRRrRb+bnZekARHq/rOHDmjTTs3aWL/hNIH0tp2/zZNfGxCWz6yRRt3btTY9jGtn1yv0dSo1k2sU3xNXE8ffrqtNqB7/CYAej2N0vL6otpOv6IYGA/ap4P+LAAAALCyMV0VAABABFUXEa8mHBpNH9XqvNp/m12nXt31ypw/cV7JLUmN7x7X5g9tVmp/SmPbx1S2ysrdyCk2EpNhGKo4FZWtspyiIztvq3inqOceek7H/vRYR/3SS53Mw88c/v3T777vd3KIdw4AAACrDUkOAACAiGo0AmP5fr/ntXO80bHLFy5r6vCUkluTWje5TmPbx7RhaoOK80WVrbLsvC0rYykxmvC2ZELxZFwjG0f02rOvdS3J0emC2H4Cw50Ek3sdiOYX9XeF1fd++rTfCZaotMGPbr2jV0951261tkgn318AAABEC0kOAAAA+DJzaUbxtXElRhOKr43LjJsqW2UV54tyio7cyuKokiFDZsyUGTdlJkzFEjFvG4lp9tVZpfalOmpHkEWjm5UNUqZ2v591OYKUDUMUAt3Ngs7dDB53q++DLHTeLb3sz27p1iLu1XcuzDqj8H0CAADAe7EmBwAAQMTce++9mpmZWdqqavfV7v/e97639PmFF17o2ufpS9OSIRmmIbfiys7Zyt3IKTOdUf5mXlbWklN0VHEqciuu5C5OlWVKxpBXZvqV6cD90i1+A8PVhZCDLIjcSdl2RfmX/NXAc7MAdNjC6PtOElrd0moh9ygnPKL8ji43SG0FAABYrRjJAQAAECFPPPGEpqenG66VUU/t/m5/litV7IrsrK3C7YJiIzHZeVuGYcgpOirdKcnO2XKKjsp22Ut4lF0v6RGSRsHGZlPUNCsbtFyraXHCKNuuqAZkGy1M3e0Fq8Po+3YSHFHo9+r9RVUv+qpZHWE/ewAAAPQfSQ4AAICISafTbe3fvXv30uejR4927XP6gFe/lbVUmC9oeG5YhmHIylgyhrzFxu2crdKdkqycJTv/7mSHYRpKH6x/D2EImjjoVsKh1wYhIBvlttXTbp82Sy4MwvPptn4nOKr7/XzfeV4AAACDgyQHAAAAfJnYPyE7b6s4X1T+Zl6x4ZgqTkWJ0YTMmCm34sopOrJylkoLJVnZxURH3pFT9LbUfZ2tx4H6BjkgG9W2R7Vdg2qQ+nOQ2goAAADW5AAAAEAb9jy4x1uDYy6v7GxWmemMt81klJ3NKjeXU/5mXoV3CiotlJZGdRQXitr70N5+N78tnQQ4exkcbTcg22qKqKgEeIO2M4x2B+nTRtvyc7qt2WiSTvs06DsTlXfKj0FqKwAAADwkOQAAAODbiWdPyM7bS0mNzHRGmeteoiM7m1Xu7Zzyc3kVbhVUuO0lOooLRakiHTt3rKttaxWAbbdcq3P8BkM7Keu3XZ0uqN3o77B0mlAJ2s4gfT8IQW4/73qv+tTv96vX/RnkvweD8OwBAADwXkxXBQAAgLacfPGkzj5wVplrGdl5W/FkXLFETDIlt+yqbJfl5J2lERyu6+qxlx4Lrf5WQdVGiw230myRar/XCKusH7XX9PPr/eX7mrWtVQKg1f6gz6BeO4K0M2jfd9KnQQXt06DvVLf6tNUzb7c/g/aL32ffrD979ewBAAAQDkZyAAAAoC3pg2k98pVHlL2Z1fyb80tTVmVnskujOXJzOWVmMrKzth69+KhS+7u/Fkej6YBaBSVbTSPULOjr59pBy3Zbs7b1sq5O+rBbdUZdq/e1H30aJc36YCU8fwAAALwbIzkAAADQtslDk3q89LgufO6CXv/y64qNxORWXEmSYRpyio72PrTX9xRVfoKO/VwjI4rrc4Rx3Xav0a9+CFq2l/cX9Jq9urd+1Nvv702v2goAAID+IskBAACAwI4/c1zHnzmu2VdnNf3KtCRvpEfqvu6P3AAAAAAAgCQHAAAAOpbal1JqH4kNAAAAAEBvkeQAAAAAIqCdhaSZWgcAAAAAPCw8DgAAAAAAAAAABhIjOQAAAIAIYHQGAAAAALSPkRwAAAAAAAAAAGAgkeQAAAAAAAAAAAADiSQHAAAAAAAAAAAYSCQ5AAAAAAAAAADAQCLJAQAAAAAAAAAABhJJDgAAAAAAAAAAMJBIcgAAAAAAAAAAgIFEkgMAAAAAAAAAAAwkkhwAAAAAAAAAAGAgkeQAAAAAAAAAAAADiSQHAAAAAAAAAAAYSLF+NwAAgKgwDGPps+u6gc4Leqz2nGZ1AwAAAAAA4C5GcgAAoLsJiGqCoTYh4fe8aoKiui0/1ur6jeoEAAAAAABAfYzkAABgUTUBsTxB4fe8ViMwml2/miDxk+i4/jfX9eY33tRb33pLkjT1o1N6/5H3a/Ljky3LAgAAAAAArCQkOQAA6DO/U1TNvjqrZ088q9xcTpIUXxOXXOnNr72pF8wXtG7rOv3M//kzumfvPd1uMgAAAAAAQCSQ5AAAoAv8Ji78nvfy776sv/xf/1Ibd2zU1CenlBhNyDANOSVHVsZScb6o7NtZffHjX9QD/+EBHfilA2HcBgAAAAAAQKSR5AAAIGTtLh7eaOqqqm//79/WN5/8prZ9Yps27dyk0YlRxdfEVbErsrKWCrcLyt/MK742rviauC7+5kU5RSe0+wEAAAAAAIgqkhwAACzyuy5Gs/OaJTjqlVt+7vLyd67d0fP/+nlt+5FtSv1wSps/slnr0+tlxkyVMiUVbhUUT8Zlxk3JkNyyq4pT0V/+L38p/ZqkZBsdAAAAAAAAMGBIcgAAoLuLgVcTELWJhtrEQ6vzav+tPd6sXDPPPfKc1r9vvTbu2Kjx3eNK/XBKGz+wUVbOUm42JzNmqlKuqGyVVS6WZedtWXlLxYWiyhfK0s8F7ZGVwe/C8K0ST0GOAQAAAACA7jP73QAAAKLCdd2lbfl+v+ct3/yUa1SXnbP11ktvaf229Vq7Za3Wblmr0a2jWrNpjYbXDSs+GlcimVA86f0bWxvzpqxaG9fw+mHpTUmreNaq5QmlRiN0mp1XTXBVN7/HAAAAAABAbzCSAwCAiJq+NK3YsJe4iA3HZBiGrKyl3I2cylZZFbtyd4SIaciMmRqKDy1tRsyQO726RxcsH0nT7nl+E1J+zVya0fSlaUlS+kBaE/sn6p63/am2Ly1Junqqddmrpxofa1S2WRk/1+h2na10Wke799SoXK/r62adYT632mu1Kt9un3byfWhUvhvPIux3tNN+8XOdTt43AAAAIEyM5AAArGrXr1/Xa6+9pu9973tLW1Xtvm9961tL+//kT/6kJ5/f/ru3ZZreWhtlu6xStqTcjZwy0xnl5/Iq3SnJLthewqNckSqSXEmGvKSHacqYZXRBp6rTjLW7oHyt8yfO67RxWmc+dUZf/Y2v6qu//lWdOXxGp43T+s///D+H3OLmGgU3mwU9tz/VOija7Jxu1elHGPfVjeu2W64b7QxaZ5j37vfcsN6HdvTqWXTSn/3ql2b30uv2AAAAAIzkAACsal/60pe0ZcsW/ezP/ux7jm3dunXp89q1a5c+f/zjH+/J5zUb16hSrsgpOrIylgo3CxqKD8nKWTJMQ+VSWaVMSXbOllNw5FiOyk5ZruPKrSxOo7RmdY/kCEOj9VlqNdp/7aVrOnPkjJKbk5o6PKXEaEKGaahiV2RlLRXni7py8YqeHH5SJ188qfTBtKTGv4auBg/9/Fq63jnV8tufanyNoOX8lPd7frt1thK0jkYjDPy2rXqs3aDv8mt2u5316lx+XT/nt1tnEGH1aStB+zRouU77s91+8TNCo537W368W88fAAAAqIeRHACAVe/GjRtav3790lZVu29ycnJp/86dO3vyOX0gLdd1ZWUtFd4pKDeXU3Ymq8x0RtmZrHI3circKqg4X5SVtWTna5IdVlmVSkXGJCM5uq1ZguPcZ89pYv+EdnxmhyYPTSp9IK3UfSlt+fAWbdq1SWM7xjS+e1zJrUmdfeCsrr98vattbRUkbSfxUatV8iXIdcMKkIZRx/Lz/CQMggbV2wmAtzov6s+t3aRdP4Lm7fZpkHKd9GdU+qXVfgAAAKCbSHIAABBRm3ZuUmwkpuLtovI388rOegmOzPWMMjMZZd/OKn8zr8I7i4mOjCUrZ8nJO3IKjhSXtLHfd9Ff1fU1Wi0K3ug8P+UaTWF15sgZbdq5SRP7J5Q+kNa2+7dp4mMT2vKRLdq4c6PGto9p/eR6jaZGtW5ineJr4nr68NN+bw1d5jcB0O8plKLaTr+iGBQP2qeD/iykaLcNAAAAaITpqgAAiChjyNDBXzqov/ndv1F8NO5NUWWVlUgmZMa93ylU7Irsgi0ra6m0UJJ1x/I+Z0vSIck1Vu90VUuLsi8mKhpNO9XsvGYLkddLjFSPnz9xXsktSY3vHtfmD21Wan9KY9vHVLbKyt3IKTbiLSRfcSoqW2U5RUd23lbxTlHPPfScjv3psbC7o2va+TU+oqPfz63fySHe1+boHwAAAAwSkhwAAETYp3/70/rOH39HmWsZyZWcoqNEMqGhxJAkqVKuqFwqy87ZKmVLKi2UVLhdUPKepBb+yYK3EPkq1miUxfL9zRYU93uNWpcvXNbU4Skltya1bnKdxraPacPUBhXniypbZdl5W1bGUmI04W3JhOLJuEY2jui1Z1/rWpIjaGC5nYB4WMHkXgTB+dX6Xb18bv1OsESlDX5E7R29euru4uLN1uSIer8CAABgZSHJAQBAxP3K67+iL3zoC5q/Oq9kIanE2oSGhoe8kQCVytJoDjtnq3C7oMT6hH75tV/Wv/sP/67fTV+VZi7NKL42rsRoQvG1cZlxU2WrrOJ8UU7RkVtZHDkyZMiMmTLjpsyEqVgi5m0jMc2+OqvUvlRH7QiyaHSzskHK1O4PGvTsRbA0CgHZZsHjbga6+/XcetnnvezPbulFf/l95rXvaqPjAAAAQC+R5AAAIOJGNozo177/a/riwS/qzlt3ZMZNJZIJGUPelEsVuyI7Z6tSqWjjBzfqF17+haWRHqvVzMzMu/6emJjwtX9kZER/93d/pyNHjkiSXnjhhbY/j353VDIkwzTkVlzZOVu5GzmVrbLcireQvFN0VHEqciuu5C5OeWV6U5S5FVfTr0x3nOQIW6Nfbi8XRkC5F78Gj/IvzvsRhO/Vc+v1vTVayL3aDr/vdT9E+R0FAAAAooQkBwAAAyC+Jq5fef1X9Le//7f669/5a71z5R3F1nr/Z9wpONr4gY365P/4Se3/xf19bmn/nT59Wrt27dLRo0ffc8zP1FOhfHYX10vJeqNrYiMx2XlbhmHIKToq3SnJztlyio7KdtlLeJRdL+kRkkaB0WZTzTQrG7Rcq+lt6tXT6FphiWrwuNHC1N1esLpXzy1K/V6b6IiiKPXVcs3a1s47AwAAAISFJAcAAAPk4C8f1MFfPihJuvHaDUnSPXvv6WeTIumNN97Qww8//J796XS67vm1+2uTI0E+z6z3RoVYWUuF+YKG54ZlGIasjCVjyFts3M7ZKt0pycpZsvPvTnYYpqH0wfrtDEO7wetOy7VjNSc4akW5bfW026fNkguD8Hy6rR994LfOVuf14r8TAAAAwHIkOQAAGFAkN6JpYv+E7Lyt4nxR+Zt5xYZjqjgVJUYTMmOm3Iorp+jIylkqLZRkZRcTHXlHTtHbUvdFa6qqXiDB0VxU2x7Vdg0q+hMAAABon9nvBgAAAKw0ex7co8x0Rvm5vLKzWWWmM942k1F2NqvcXE75m3kV3imotFBaGtVRXChq70N7+938toQRjI1igqPVFFFRCUYHbWc/ntvVU4235ed0W7PRJJ32adB3pl/vVJSn7QIAAAD8IMkBAAAQshPPnpCdt5eSGpnpjDLXvURHdjar3Ns55efyKtwqqHDbS3QUF4pSRTp27lhX29YqANtuuVbnRCGw22kdy++rW0HhThMqQdsZ1efWKT/veq/61O/3q5/92U7dQf87AgAAAHQD01UBAAB0wckXT+rsA2eVuZaRnbcVT8YVS8QkU3LLrsp2WU7eWRrB4bquHnvpsdDqbxVsbLRocCvNFqn2e4169fn5ZX0QndTR6r5aJQBa7Q/6DOq1I0g7o/zcmtXZbP/yOoMG3bvVp62eebv92cm7FoTfdybKyS8AAACsPIzkAAAA6IL0wbQe+cojyt7Mav7N+aUpq7Iz2aXRHLm5nDIzGdlZW49efFSp/d1fi6PRdECtgpKtphFqFvQd5IBns/vqZV1+nk87+8OoM+pava/96NOoCTKCpFnfrYT3BgAAAIOHkRwAAABdMnloUo+XHteFz13Q619+XbGRmNyKK0kyTENO0dHeh/b6nqLKT/CwkwBjp8HJdsv3IhgaRh29vK9+lI3CcwuaOOj0ut0q327SIKh+3V8YdQMAAABhIckBAADQZcefOa7jzxzX7Kuzmn5lWpI30iN1X/dHbgAAAAAAsJKR5AAAAOiR1L6UUvtIbAAAAAAAEBaSHAAAABgY7SwkzXQ6AAAAALDysfA4AAAAAAAAAAAYSIzkAAAAwMBgdAYAAAAAoBYjOQAAAAAAAAAAwEAiyQEAAAAAAAAAAAYSSQ4AAAAAAAAAADCQSHIAAAAAAAAAAICBRJIDAAAAAAAAAAAMJJIcAAAAAAAAAABgIJHkAAAAAAAAAAAAA4kkBwAAAAAAAAAAGEgkOQAAAAAAAAAAwEAiyQEAAAAAAAAAAAYSSQ4AAAAAAAAAADCQSHIAAAAAAAAAAICBRJIDAAAAAAAAAAAMJJIcAAAAAAAAAABgIJHkAAAAAAAAAAAAA4kkBwAAAAAAAAAAGEgkOQAAAAAAAAAAwEAiyQEAAAAAAAAAAAZSrN8NAAAAAGoZhrH02XXdwOc1Ot6snN+6AQAAAADRwEgOAAAAREY1yVBNMNQmHdo5zzAMua67tDXaX1uu2TEAAAAAQDQxkgMAAACRUk1KtEo0NDqvmqxoVqbdY43MXJrR9KVpSVL6QFoT+yfqnrf9qbYvLUm6eqp12aunGh9rVDZIGb/X6KS9fgW5r1bl/ZRdXq7X9XWzzk77tNG1WpVvt087fb969SzCfkfbeQ6dvDf1rhHGdxYAAKCbSHIAAABgxQk67ZTfcudPnNflC5cVXxuXDEmLp9p5W3se3KMTz55ou81BbX+qfhCyWTC4emwQg5ed3lej8o36sVWdzbRqa9jtDFpnmO+K374K2qedCLtPu9GfnfRLJ+9bWG0AAADoB5IcAAAAWHHqTVG1XL39rcpde+mazhw5o+TmpKYOTykxmpBhGqrYFVlZS965uIYAACAASURBVMX5oq5cvKInh5/UyRdPKn0wLal1QNNP8LFZMLVZALPdcn6CvUHbG5ZW/ekn+Fx73E8/1pZpNwi8/Jrdbme9Opdf18/57dYZRFh92krQPg1artP+DNov9eru9jMEAADoN9bkAAAAwKrTbEqrRq69dE3nPntOE/sntOMzOzR5aFLpA2ml7ktpy4e3aNOuTRrbMabx3eNKbk3q7ANndf3l6126A0+rIGk7iY9B4Scw3O41/CQMggbV2wmAtzrP71RM7T73MPrUT/3Lr9uP97DdPg1SrpP+DNovYbxvra4DAAAQVSQ5AAAAECnVKaNaLfzt97x65RqN7GjmzJEz2rRzkyb2Tyh9IK1t92/TxMcmtOUjW7Rx50aNbR/T+sn1Gk2Nat3EOsXXxPX04afbaluUDfoUNn4TAL2+z+X1RbWdfkUxOB60Twf9WdTjp61RfIYAAADNMF0VAAAAIqO6iHg14dBo+qhm5y1fiHz5NWr/rT3erNz5E+eV3JLU+O5xbf7QZqX2pzS2fUxlq6zcjZxiIzEZhqGKU1HZKsspOrLztop3inruoed07E+Pdd45EdGLhZPRvn7/Ar/fySHer84NUrIGAACgFkkOAAAAREqjaaSarZ8R9Bp+j1++cFlTh6eU3JrUusl1Gts+pg1TG1ScL6pslWXnbVkZS4nRhLclE4on4xrZOKLXnn2ta0mOThfE7mVguBd1EqS9K6wkgJ8+7XeCJSpt8COq7+ig9B8AAEA9JDkAAACAJmYuzSi+Nq7EaELxtXGZcVNlq6zifFFO0ZFbWRxVMmTIjJky46bMhKlYIuZtIzHNvjqr1L5UR+0Ismh0s7LdnKe/0cLHvRKFQO3VU95911vsuZv90ejanQaxgyx03i39fr/CEHZ/hfG+ReF7AwAAEARJDgAAAETCzMzMu/6emJjo6v5isagdO3ZIkl544QUdOXKk7ufR745KhmSYhtyKKztnK3cjp7JVlltxZWUtOUVHFacit+JK7uJ0WKZkDHllpl+Z7jjJEbZ6wdAwNFr4uBpo7Va9Uf4lej+C8GEkAvz0aRQSWL14v8LQq3e03WcyiEkiAACAWiQ5AAAA0HenT5/Wrl27dPTo0fcca3fqqSDTXbX87EoVuyI7a6twu6DYSEx23pZhGHKKjkp3SrJztpyio7Jd9hIeZddLeoSkUWC00a+3W5X1Uy5MtYHosEU1wdFoYepuL1jdLBHg95m3k+CIQr938/0KQy/6Ksj7FqVnCAAAEBRJDgAAAETCG2+8oYcffvg9+9PpdN3zw9ov6V3JleWfZ9Z7I0CsrKXCfEHDc8MyDENWxpIx5C02budsle6UZOUs2fl3JzsM01D6YOO6O9Vu8LrdclEPgka9fVK021ZPu33aLLkwCM+n23rdB0Hq4RkCAIBBRpIDAAAAaGJi/4TsvK3ifFH5m3nFhmOqOBUlRhMyY6bciiun6MjKWSotlGRlFxMdeUdO0dtS90VrqqqVYpCDr1Fte1TbNaii0p9RaQcAAEA3kOQAAAAAWtjz4B5duXjFS2wMeQuPJ0YTMuOm5Epluyw7b8vKeomO6qiO4kJRex/a2+/mBxbm9D9hTyUUZDH0ZiNXohIEDtrOMKZr6nSB+U6u1alm995pnwZ9Z6LyTjUTpWcIAAAQlNnvBgAAAABRd+LZE7LztjIzGWVns8pMZ5S5nlFm2vs793ZO+bm8CrcKKtwuqLRQUnGhKFWkY+eOdbVtjYK7rQLe7QTEO522qHZ/GAHTToOvy9vZ7bVCGu1v1f6g7Qyy9sIgBLTDeL/C6lO/369e9men7xsAAMCgYiQHAAAA4MPJF0/q7ANnlbmWkZ23FU/GFUvEJFNyy67KdllO3lkaweG6rh576bHQ6m8VVG32S/N2y3Wi24s/116/1a/36+2rlqlXtlUCoNX+oM+gXjuCtLNVuUY66dOggvZp0PerW33a6pm325+dvGut6gMAAFipGMkBAAAA+JA+mNYjX3lE2ZtZzb85743mmM4oO5NdGs2Rm8spM5ORnbX16MVHldrf/bU4rp5qHNAPUq4qyK+/m53bqr5eaRbM7mVdfp5PO/vDqDPqOn2/utGnUbKSnz0AAEAzjOQAAAAAfJo8NKnHS4/rwucu6PUvv67YSExuxZUkGaYhp+ho70N7fU9R5Sfw2Elwsh9lux1MDeP67V5jpT+DXiZ4Oq2z07b2ot5+vS9hlO/WtQAAALqJJAcAAADQpuPPHNfxZ45r9tVZTb8yLckb6ZG6r/sjNwAAAAAAd5HkAAAAAAJK7UsptY/EBgAAAAD0C0kOAAAAAH3TzkLJTJ8DAAAAYDkWHgcAAAAAAAAAAAOJkRwAAAAA+obRGQAAAAA6wUgOAAAAAAAAAAAwkEhyAAAAAAAAAACAgUSSAwAAAAAAAAAADCSSHAAAAAAAAAAAYCCR5AAAAAAAAAAAAAOJJAcAAAAAAAAAABhIJDkAAAAAAAAAAMBAIskBAAAAAAAAAAAGEkkOAAAAAAAAAAAwkEhyAAAAAAAAAACAgUSSAwAAAAAAAAAADCSSHAAAAAAAAAAAYCCR5AAAAAAAAAAAAAOJJAcAAAAAAAAAABhIJDkAAAAAAAAAAMBAIskBAAAAAAAAAAAGEkkOAAAAAAAAAAAwkEhyAAAAAAAAAACAgRTrdwMAAAAADAbDMJY+u67b9nm1+1sdX359v3UDAAAAWF0YyQEAAACgpWqSoZpgqJewaHWe67rv2paXqz1WW67ZMQAAAACrGyM5AAAAAPhSTUy0SjT4Oa+auOiWmUszmr40LUlKH0hrYv9E3fO2PxXs+ldPtS579VTjY43KNivj5xrtlF9+jXbLtrpeLb/XDnpPy8v1ur5u1hnGu1LvWq3Kt9unnXwfGpXvxrMI+x3txXc2jO86AAArGUkOAAAAAJG2PFnSLDly/sR5Xb5wWfG1ccmQtHiqnbe158E9OvHsiS639q7tT9UPRDYLBlePBQnSNquz3esE0a37anZPQe+hkz4M0s6gdXbap36vFeS8MIXdp93oz076pRv1tvNdBwBgpSPJAQAAAKCn6o3iaGfUR73y1166pjNHzii5Oampw1NKjCZkmIYqdkVW1lJxvqgrF6/oyeEndfLFk0ofTEtqHVj0E0RsFkxtFogMWs5P+X5p1Z9+gs+1x/32R/VYu/e//Jrdbme9Opdf18/57dYZRFh92krQPg1artP+bLdfWo2eaVZvGO8bAACrAWtyAAAADCDDMJa2IOe1Uz5o3UC7Gq3X0cq1l67p3GfPaWL/hHZ8ZocmD00qfSCt1H0pbfnwFm3atUljO8Y0vntcya1JnX3grK6/fL1Ld+FpFSRtJ/FRq1Xypd1f9YcZIPUTGG73Gn4SBkGD6u0EwFud163nFkaf+ql/+XX7EThvt0+DlOukPzvtl06+s0H7BgCA1YIkBwAAwIAJcwFoP/Us38cC0KtX9Xn7TY4FfT/aXa/jzJEz2rRzkyb2Tyh9IK1t92/TxMcmtOUjW7Rx50aNbR/T+sn1Gk2Nat3EOsXXxPX04acDtW0liUqg1G8CoNejVJbXF9V2+hWV510raJ8O+rPwYzXcIwAAYWG6KgAAVgG/c9k3O6/RsXpBzGbB90Zlu7kA8UoU5gLQ9VSDzGElMW69cUuSNL57PJTrofeq78Py5Jn07qREs/OWn7t8f21d9equd/z8ifNKbklqfPe4Nn9os1L7UxrbPqayVVbuRk6xkZgMw1DFqahsleUUHdl5W8U7RT330HM69qfHgnZJz4U1+oKgaG91Y9RMkPr7VV8UkysAAGBlIckBAMAKVxtorAYemwUY653X7JjfZMjyY37m2Ed/hPUsXv3iq/r273xbt/7hlmIJ7392Opaj8Z3j+pH/6Ud038n7Oq4DvdXovaiXlOj0Gn6PX75wWVOHp5TcmtS6yXUa2z6mDVMbVJwvqmyVZedtWRlLidGEtyUTiifjGtk4oteefa1rSY5OF8T2ExgOEkzuV8CdxMpdYSUB/PRpvxMsUWmDH916R6+e8q7dam2RqPcPAABRRpIDAIBVIIxf/fu5RrPgeCeB88LNgi598ZKu/MUVzf1/c5KkLR/eog/+xAd14BcPaGTjSKDr4r1aPSc/ozuckqMvffxLWvjHBa3ZuEaTByZlJsylBaBzczl99Te+qpe/8LJ+4eVfkDHElFcIZubSjOJr40qMJhRfG5cZN1W2yirOF+UUHbmVxcTskCEzZsqMmzITpmKJmLeNxDT76qxS+1IdtSPIotHNygYpU7s/yMLXvRKFQG6zoHM3kzGdPrdGovS8e9mf3dKtRdyr71yv6gQAYDUhyQEAACLtW5//lv7q3/6VJCkxmtDaDWtVqVQ09//O6frL1/Xiv31RR544ok/8xif63NKVo94i5bWJj2bTlZUyJX3h3i9oKDGk7T+6XaOpUcWTcbkVV07BUXG+qOSWpHI3c3rnB+/od6Z+R7/6xq9294bQsZmZmXf9PTEx0df9//AP/6CdO3dq+tK0ZEiGacituLJztnI3cipbZbkVV1bWklN0VHEqciuu5C6+s6ZkDHllpl+Z7jjJEbZGv/hert2Acr8CzlH+pXo/+iSMRICfPu31vTVayL3aDr/vdT9E+R0FAACtkeQAAAChaHcUh58RAef/+Xl9/+L3lfqhlJJbk0qMJiRDcgqOSpmSCrcLKtwq6Ou/9XXNXprVT//JT4d2P1Hnd82MdtfW8Lt+QqNjv//R31c8Gde2+7dp/N5xrUuv01BiSE7RS3Dkb+a9X9wnTBmGoYV/XNAf7P0D6VFfzUMfnD59Wrt27dLRo0ffc6zdqae6st+VKnZFdtZW4XZBsZGY7LwtwzDkFB2V7pRk52w5RUdlu+wlPMqul/QISaPAaLMpapqVDVrOzwiFfk1TFbXgcaNFm7u9mHO7z62edhIcUej32kRHFPWir5rV0c6zBwAA9ZHkAAAAfdNsRMCXj39Zb33jLW3/1HZt2rVJ6ybWKTYSU9kqq3SnpPytvIbXDSs2EpM5ZOrv/8vfq/xQWfpwr++i98JYAHr5+ijLjzfSbM2V5//18yrdKekDn/mAtuzZotQPp7T+fevlVlwVbhWUvZGVGTO9oHR1EeiSo4WrC9I3JB1prx/QO2+88YYefvjh9+xPp9N1z+/2/l27dnnHD3jHraylwnxBw3PDMgxDVsaSMeQtNm7nbJXulGTlLNn5dyc7DNNQ+mD9OsLQbvC603J+NAs2hx3sjVKgvZEot62edvu0l897EPU7wVHdT6IDAIDOkOQAAGAVCONX/+2OCAjSvqrv/8X39f2/+L6mDk/pnr336J6P3qP1k+slUyotlJSfyy8lN1zXleu4KttlvfF/vSGNSHp/6E2MnE4XgPa7Porf61Wcil75g1c0vnt8afHn8XvHtfEDG1W4XZBhGl5SI+/IznkLQZcyJSVGExreMCz7ZVvGp1ibA+2Z2D8hO28vjRKKDcdUcSpKjCZkxkxvmrSiIytnqbRQkpVdTHTkHTlFb0vdF62pqlaKQQ6gR7XtUW3XoKI/AQBYOUhyAACwwoXxq/9mx5ZfZ7lGx5qNCPiL3/wLbXz/Ro1tH9OmD27SPR+9R+O7xlV4p6DsTFZypbJdllN0ZOdtL2ies1SaL8n+qi39ou/uQUje+f47cvKORjaOaHjdsOJr4zIMQ3be9hZ/Ng1v0ee4qaHEkIaGhxQbjik2HFN8TVy6KbnvhDd9EFaPPQ/u0ZWLV7zExpC38HhiNCEzbi79t8LO27KyXqKjOqqjuFDU3of29rv5belk2p9eTm3U7vVa/ZI9KsHooO0MY7qmIH0a1rU61ezeO+3ToO9MVN6pZgblewEAQBSY/W4AAADoPtd1l7bl+/2c5+dYs7rbaVPuRk43v3tTa7eu1cjGES9ovmHYC44PD2loZEixNTHFRmLevzWf46NxaU4yCit3REAmk3nXVpXP53X16tWlv3v9eebSjGRIQ/EhSZJTcpS/lVdmOqPCrYLsnL20GLTrukuLRRtDhsyYKdMwpen2+wM48ewJ2XlbmZmMsrNZZaYzylzPKDPt/Z17O6f8XF6FWwUVbhdUWiipuFCUKtKxc8e62rZGwd0wFgivd04Ugp6dtmH5fXVrLYdWz6ZV+4O2M8hzi8JzbcXPu96rPvX7/erX+jR+9zc7J8prnAAA0A+M5AAAAJEyc2lGZsxUbCSmofiQ3LKr0nxJGSPjreNQ9ObSl+uNBjHMxSD5kDdKwBwyVb5e7vdtdMUf/dEfaceOHUql7k6vs27dOknS3Nycvv71r+vnf/7nJannnz+S+YgMc3ENhIKt4jtFZWezSwtA2wVviiqn4KhslVWxK6qUK1LFS3iZMlXJV8LuMqwSJ188qbMPnFXmWkZ23lY8GVcsEZNMyS1709k5eWdpBIfrunrspcdCq79VwLHZr7DbLVc7KiBqgc7a9rT69X69fc3uq1UCoNX+oM+gXjuCtDPoc+ukT4MK2qedjDLqRp+2eubt9mfQfvH77MP8XgAAsNowkgMAgBXq/vvv10/+5E/qO9/5jr7zne/o+vXr+vznP790PKqfb79xW+aQKVW80QClOyVl3/Z+nZ27kVNxoejNqV/yFg92y3dHgxiGIdMwZdxamSM53nzzTT3//PPavXv30la1ffv2paSDpJ5/3vpDW+VW3KVpw3JzOWVnsku/ps/P5VV4p6DSnZLs3OLiz1bZe4aOq7JblruV6aoQTPpgWo985RFlb2Y1/+a8N5pjOqPsTHZpNEduLqfMTEZ21tajFx9Van/31+K4eqpx4DJIuVblW5WLumb31cu6/DyfdvaHUWfUtXpf+9GnUdKsDzr5vgMAAA8jOQAAWKEeeOABlct3RzQYhqFTp+7+f8RR/bzh/Ru80QBFW6U7JeVv5mXGTFlZa2nxaitrycp6owKc0uLIAMcbGVBxK3LHCJb32sTHJlS2yt7C8Lfyiq2Jya24SmQWF4AuewtAlzKLayJUF4Aues/QLbsyJldmcgq9MXloUo+XHteFz13Q619+XbER7x2UvKnRnKKjvQ/t9T1FlZ8AYidBxk4DlGEGOMO6VhjXafca/XoGQcv28v6CXrNX99aPevv5ne3kGiQ0AABojiQHAAAr2NDQ0Lv+HhkZifzniY9NyC27su5YKtwuKL427gXL13mLClfKFTmFu8FyO2e/O9lRqRAs74PEaELv+8T7dOt7tzS8blhmzFTZLiuRrFkA2vIWgC5lSiotlGRlLFk5L2GlKcmNkZxC544/c1zHnzmu2VdnNf2Kt9BL+mBaqfu6P3IDAAAAQO+R5AAAAJGyftt6bdi+QflbeQ1vGNZQfEgVp7I0IkCuN42VnfNGerxrVEDekcYkd5RgeT/89Nmf1u/t/j1lb2Qlw3tOiWRCQwkv2VZxKnKK3roIpTt3Ex1O0ZGO97nxWHFS+1JK7SOxAQAAAKx0JDkAAEDkfOq3PqU//6U/V2I0IcMw3hMsL9tlOYXFYPnCYqIjY8ku2NJP9rnxq9iGqQ361BOf0ou//aLciiu7YN99bsbiAtBW2VuEPLuY6MiU9Ol/82ldzF2UVkhuyjDujiSqrhfT7nlBjtXub1U34Ec7C0kznQ4AAAD6hSQHAACInB965If0X//wv2rm1Rm5riu7+N5guVNyZOcXg+ULJRVuFbTtE9v0gz0/WDHB8kF0+H87rPhIXM//q+dlZSwNrxvW0PCQDNOQ67qq2IujOTKW7KKtn/j3P6GDv3pQF09f7HfTQ1FNNLiuK8MwZBhG3WRDs/OWl/F7rHo9AAAAAFhNSHIAAIBI+vkXfl5f+sSXNPfanOx77iY5DNOQW3GXRnPYOVuF+YLSH0vr5772czp9+nS/m77q3f/r92vqk1M6/zPnlZ3NquyUNRRfHIVjeZ+Tm5P6ub/8OaX2r7zphKqJhmoCo93zmiUqwk5izFya0fSlxXUrDqQ1sX8i1OtjsDE6AwAAAIOAJAcAAIisf/Htf6E/+6U/0+VnLys3m1NsTUxDI0PeItZFb9qj2JqY9p3cp3/6e/+0381FjfShtE59/5Te+tZb+sHXfqB/fOkfJUnv++T7tOPTOzT1o1N9buHgCjrNVa3zJ87r8oXLiq+NS4aWRj/ZeVt7HtyjE8+eCLXNAAAAANAtJDkAAECk/dQf/JQO/cohffv/+Lbe+qu3NH91XpI09v4x3Xv4Xv3Ir/+Itnx0S59biboMaepHp0hodKDedFeNpquql/BYvu/aS9d05sgZJTcnNXV4ylv3xjRUsSuyspaK80VduXhFTw4/qZMvnlT6YLpLdwYAAAAA4SDJAQAAIu+eH7pH/+zpf9bvZgA91Wg9j6CuvXRN5z57ThP7JzS+e1zJrUnF18a9ReKz3rRv+Zt5JUYTysxkdPaBs3rkK4+EVj8AAAAAdANJDgAAACBE1eREs/U4Wp3XbMHyoImPM0fOaGL/hCb2T2jzhzZr3eQ6mXHTW9fmdkHDc8OKDcdkDpmSpMy1jJ4+/LT0LwNVBwAAAAA9QZIDAAAACEk1aVFNXDSbWqrZebX/1h5vtkj58mRJ7bHzJ84ruSWp8d3j2vyhzUrtT2ls+5jKVlm5GznFRmIyDEMVp6KyVZZTdGTnbRXvFGWdtySW6AAAAAAQUSQ5AAAAgBA1GmnRbG0NP/s7KXf5wmVNHZ5ScmtS6ybXaWz7mDZMbVBxvqiyVZadt2VlLCVGE96WTCiejGtk44isy1bT9gAAAABAP5n9bgAAAACA7pm5NKP42rgSownF18Zlxk2VrbKK80U5RUduZXFUyZAhM2bKjJsyE6ZiiZhiiZgUk4zZ5lNvAQAAAEC/kOQAAAAAQjAzM/Ourdn+F154Yel4tz9/45lvSIZkmIa3yHjOVu5GTpnpjPI387Kylpyio4pTkVtxJXdx6itTXuLDMKXpoL0CAAAAAN3FdFUAAABAh06fPq1du3bp6NGj7zlWbxqp2n3d/uztkCp2RXbWW2Q8NhKTnbdlGIacoqPSnZLsnC2n6Khsl72ER9n1kh4AAAAAEGEkOQAAAIAQvPHGG3r44Yffsz+dTr9nX20ypOufHz6qH/z+D2RlLRXmCxqeG5ZhGLIylowhb7FxO2erdKckK2fJzi9Ldriu3DTJDgAAAADRRJIDAAAAWMEm9k/IztsqzheVv5lXbDimilNRYjQhM2bKrbhyio6snKXSQklWdjHRkXe8NTscV0r1+y4AAAAAoD6SHAAAAMAKt+fBPbpy8YqX2BjyFh5PjCZkxk3Jlcp2WXbelpX1Eh3VUR3FhaK0p9+tBwAAAIDGSHIAAAAAK9yJZ0/oyeEnlZnJSJKcoqNEMiEzYcowvCmrnKIjO+9NW1VaKHkJjoqkE5KYrQoAAABARJn9bgAAAACA7jv54knZWVuZaxllpjO6M31H2emsMjMZZWezys3llJ/Lq3C7oPztvFzX1WMvPdbvZgMAAABAUyQ5AAAAgFUgfTCtR77yiLI3s5p/c16ZaS/ZkZ3JekmOt3PKzeWUmcnIztp69OKjSu1nMQ4AAAAA0cZ0VQAAAMAqMXloUo+XHteFz13Q619+XbGRmNyKNxeVYRpyio72PrRXx84d63NLAQAAAMAfkhwAAADAKnP8meM6/sxxzb46q+lXpiV5Iz1S9zFyAwAAAMBgIckBAAAArFKpfSml9pHYAAAAADC4SHIAAAAA8MUwjKXPrusGOi/Isdr9reoGAAAAsLqw8DgAAACAlqqJhmqCYXniwc95hmHIdd2lze+x6vWqGwAAAABUMZIDAAAAgC/VBEO9JISf83qZoJi5NKPpS4vrjRxIa2L/RN3ztj8V7PpXT7Uue/VU42ONyjYr4+cafsoHLedHp/cV1j31ur5u1hnGu1LvWq3Kt9unnXwfGpXvxrMI+x3ttF86uU5Y31sAAAYdSQ4AAAAAfdcqIeI3WXL+xHldvnBZ8bVxyZC0eKqdt7XnwT068eyJUNvdzPan6gchmwU9q8eCBGmb1dmqbKtyfnTrvpq1LWiSqh99GKTOTvvU77WCnBemsPu0G/3Zj35pVW8Y31sAAFYCkhwAAAAAeq46PZWfv+slPJbvu/bSNZ05ckbJzUlNHZ5SYjQhwzRUsSuyspaK80VduXhFTw4/qZMvnlT6YFpS64CmnwBis2BqsyBk0HJ+ytfT6Ff87dTZSqv+9BN8DtK26rF2A9HLr9ntdtarc/l1/Zzfbp1BhNWnrQTt06DlOu3PdvvFT5Km0//OAACw2rEmBwAAAICeqpek6MS1l67p3GfPaWL/hHZ8ZocmD00qfSCt1H0pbfnwFm3atUljO8Y0vntcya1JnX3grK6/fD20+utpFSRtJ/FRq1VQNEiwNKwAuZ/AcLvX8JMwCBpUbycA3uq8bj23MPrUT/3Lr9uP0QFB38t2ynXSn/3olzC+7wAArAYkOQAAAAD4Up0yqtl6HK3OCzvBIUlnjpzRpp2bNLF/QukDaW27f5smPjahLR/Zoo07N2ps+5jWT67XaGpU6ybWKb4mrqcPPx1qG6LOb7C0n1MVRa1ty+uLajv9imJAPGifDvqzkKLdNgAABg3TVQEAAABoqbpmRjVx0WxqqWbn1f5be7zZmhzLkyW1x86fOK/klqTGd49r84c2K7U/pbHtYypbZeVu5BQbickwDFWcispWWU7RkZ23VbxT1HMPPadjf3qss47poXbXYEA09Pu59Ts5xPvaXBhT5gEAsNqR5AAAAADgS6MRGMv3+z0v6PVrXb5wWVOHp5TcmtS6yXUa2z6mDVMbVJwvqmyVZedtWRlLidGEtyUTiifjGtk4oteefa1rSY5OF8T2E9gcpGAyv1q/K6zn5qdPoxAoj0Ib/Ij6OzpI33cAAHqNJAcAAACAgTRzaUbxtXElRhOKr43LjJsqW2UV54tyio7cyuKokiFDZsyUGTdlJkzFEjFvG4lp9tVZBV3wKwAAIABJREFUpfalOmpHkEWjm5UNUqZ2f5SDn1Fo29VTXl/VW2C6m4Hubj23IAudd0sv+7NbetFfnSQzg1wDAICVjjU5AAAAECnVqY78rPtQ77x2ysOfe++9VzMzM0tbVe2+5fu/+c1vLv39wgsvdOXz9KVpyZAM05BbcWXnbOVu5JSZzih/My8ra8kpOqo4FbkVV3IXn7spGUNemelXpjvpmq7wGxiuLoTcr4Wi/YpyMLaa7KhuvRDGc/PTp71OMLRayD3KCY8ov6NVg/J9BwCgH0hyAAAAIDKWr+PQKBHR7DzXdVtOi0SCw78nnnhCR44cWerX2r6t3Vdvf+3fXfvsShW7Ijtrq3C7oOxsVpnpjHJv51S4VVDpTkl2zpZTdFS2y17Co+x6SY+QLA8+1gYhWwV2OynXaF+UgslRDR43ChR3O4AcxnNrJ8ERhX6PQhuaiVJfNTIo33cAAPqF6aoAAAAQKY0Wog563nLVRbJJdPiXTqfb3l977OjRo135PLPeGz1iZS0V5gsanhuWYRiyMpaMIW+xcTtnq3SnJCtnyc6/O9lhmIbSB+vfQxiaTYvUjXJRM6jB4yhrt0+bBcAH4fl0Wz/6gH4HACB8JDkAAACwalQTHH68c+Ud/e0X/lY/+PoPdPvKbUnSpp2btOOf7NCh/+GQxt4/1s2mwoeJ/ROy87aK80Xlb+YVG46p4lSUGE3IjJlyK66coiMrZ6m0UJKVXUx05B05RW9L3dfZehyob5ADuVFte1TbNajoTwAAVg6mqwIAAMCq0E6C4+L/fFF/ePAP9eqZV1WcL2rj+zdqw/s2KP92Xpf+8JL+077/pK/9y691ucXwY8+De7w1OObyS1NVZaYzysxklJ3NKjeXU/5mXoV3CiotlJZGdRQXitr70N5+N78tnQRjW01tE2bAt91r9bJtnQjazn71abNp1GrP6bZmo0k67dOg70y/3ql2ppbq9/sOAMAgIckBAACAVWP5ouT1pqz64x//Y736pVeVui+lnf/NTn3gxz6gyUOTSu1LKfWxlFI/nNLo1lH9ze/+jc79t+d6fQtY5sSzJ2Tn7aWkRmY6o8x1L9GRnc0q93ZO+bm8CrcKKtz2Eh3FhaJUkY6dO9bVtrUKwLZbrtU5fgO3y8uGOad/p8HjbrbNz3W73YdBnltUkjzN+HnXe9Wnfr9f/ezPduvu5PsOAMBqwHRVAAAAiBS/a2a0u7bG8lEc9UZ2nH3grGb/n1ltP7pd47vGNToxqthITGWrrNJCSflbeeXX5TU0MiQjZujqi1d17qfOSQfbu0eE6+SLJ3X2gbPKXMvIztuKJ+OKJWKSKbllV2W7LCfvLI3gcF1Xj730WGj1B1kk3E8At9Fiw9Wy7SYAWpXtNFhae00/v94Po21+E0lBn0G9dgRpZ9Dn1kmfBhW0T4MmpLrVp62eebv92cm7FlQn33cAAFYTRnIAAAAgMqpJh2riojYJUZvMaHVe7UgNv0mQv7/w97r219c0eWhS93z0HqUPpLXt49uUui+l8d3j2rB9g9ZPrldya1LJLUmtHV+r0YlRvfmNN6XvBr9ndC59MK1HvvKIsjezmn9zfmnKquxMdmk0R24up8xMRnbW1qMXH1Vqf/fX4mg0HVCrIGiraYSaBX39XLud/b3Uy7b1ow87qTPqWr2v/ejTqAk68mIlvzcAAISFkRwAAACIlEbrZizf7/c8v/V8/be+7q29MbVBmz64SVs+ukXju8dVuFVQZjojt+KqbJVlF2zZeVtWzpKVtVRKlmR/zZZ2+6oWXTJ5aFKPlx7Xhc9d0Otffl2xkZjcymIyzDTkFB3tfWiv7ymq/AQPw1gjox/luxUYDeO6YQWAo1q2l/cX9Jq9urd+1Mt3FgCAlYkkBwAAAFa97ExWt6/c1vbD27Vm4xoNjw0rkUxIksyYqaHhIcVGYu/d1sQUT8altyQj52/ECLrr+DPHdfyZ45p9dVbTr0xL8kZ6pO7r/sgNAAAAAL1HkgMAAACr3vSl6aVkhjFkqOJUVJwvSoZUcSpyio4qTkVyF6fAMg2ZQ6bMmLcZQ4bcaX8jSNAbqX0ppfaR2AAAAABWOtbkAAAAQN8dPXpUn/3sZ3Xp0qWlrer5559f+nz69OmufJ7//rzMIdObkqrkLTKenc0qM51R/kZepfmS7Jwtp+iobJflll1vKqTFpMeQMSTdDqEjgAjZ/pT/DQAAAOgXRnIAAACg744cOdLw2Kc//emlz0888URXPr/xZ294IzYKjkp3SsrfzMscMmVlLZlDpspWWVbWW4PDzt9NdlSciipOReVKWdoU4MYBAAAAAB0hyQEAAIBVL/2xtCrlikp3SircLii+Ni634iqRSciMmXLLruyCLStjeVvOkpN3vGSHVZZbceWmma4KKwuLHQMAAGAQkOQAAADAqjc6MarxneMq3CwotyEnM2aqbJeVGE1oKD7kTWNllWXlLFl3FrfFUR12wZbGJSX7fRcAAAAAsPqQ5AAAAAAkffq3P60LD19QfF1chmHIKTlLSQ5JqtgV2UVbVtZS6U5JxTtFWVlLTsGRfrrPjQcAAACAVYokBwAAACDp3v/uXr3/yPt19ZtX5Va86akSyYSGEkOSIbllbzSHnV9MdCyUlL+V1wd//IP67q7vSsxWBQAAAAA9Z/a7AQAAAEBUPPznD2vqR6e08NaCstNZZaYzykxnlJ3JKvt2Vrm3c8rN5ZS/mVfuZk4f/LEP6qH/8lC/mw0AAAAAqxYjOQAAAIAaj/zfj+hr/+preuU/vqLs21kNDQ8pNuz9z2an6KhcKmtozZA++Zuf1NHTR/vbWAAAAABY5UhyAAAAAMv82L/5MR347w/o5f/4st782pu69b1bkqRNuzfpAz/2AR361UNav219n1sJAAAAACDJAQAAANSxYfsG/fjnf7zfzQAAAAAANMGaHAAAAAAAAAAAYCCR5AAAAAAAAAAAAAOJJAcAAAAAAAAAABhIJDkAAAAAAAAAAMBAIskBAAAAAAAAAAAGEkkOAAAAAAAAAAAwkEhyAAAAAAAAAACAgUSSAwAAAAAAAAAADCSSHAAAAAAAAPj/2bv/EDnOPM/zn4jKzCorS1JJKq0ys+xSe8e23K0RLVVL3tme0Umww/mYo/cGqUy7rT0M0m0fu3OcZ4+F4+YwpkHs/bfgP25nm1us5RByW0I+GIYds2bZ1uLTcNPWuRtrBdeMcGtGyiyrZLlU+SsyIjLi/sjKcjldmRkR+Suy6v2CpFOR8cTzxDejXM33W8/zAAAwlihyAAAAAAAAAACAsUSRAwAAAAAAAAAAjKXEqAcAAAAAbCWGYay/932/p/MMw/jGZ0GvDwAAAADbATM5AAAAgD5pFiCaxYeNBYmw57U75vv++qvd9QEAAABgu2AmBwAAANBHzcJFtyJEp/OaxYzW41FmbhRuFZS/lZck5Y7nlF3IbnrewbdDX1qSdO+N7m3vvdH+s3ZtO7UJco0g7aO2C6LX++rXPQ27v0H22Y9nZbNrdWsfNqa9/Dy0az+I76Lfz+ggf2Z7jSkAANjaKHIAAAAAMbLZElVRXFu8pjvX7yi5IykZktYu6VQcHX7lsBavLvbcR1AH3948Cdkpcdn8LGris12fvbQLalD3FfWeOuklFlHGGbXPXmMa9FpRzuunfsd0EPHsJS79/B4BAACaKHIAAAAAMRGmwNHu3Ps37+vSqUtKz6Y1f3JeqemUDNOQ53iyS7asFUt3P7yri5MXdf6j88qdyEnqntAMknjslEztlKSN2i5I+zDtwvQZ9tpB+2g3wyDo2JqfhU1Eh41Fr+PcrM/W6wY5P2yfUfQrpt1EjWnUdr3GM2xcus2eifrfCgAAAPbkAAAAAGLEMIz1V/Pfm53TrsBx5QdXlF3I6tnff1ZzL80pdzynzNGM9n97v/Y+v1czz85o3wv7lD6Q1uWXL+vBXz0Y6P10S5JGTWZ2K75EKdr0K4EaJDEc9hpBCgZRk+q9xCLsOKN+b/2IaZD+W687iqR62JhGaddLPHuNCwUMAADQbxQ5AAAAgD7qVJzodt7GTcU37tnR2q7dbI9Lpy5p73N7lV3IKnc8p6d/52llv5fV/u/s157n9mjm4Ix2ze3SdGZaO7M7lXwqqXdOvhP5XreyUS5V1C0JPOyxtfYX13EGFcdketSYjvt3AQAA0A8sVwUAAAD0SXOz8GbhYmMxYmNxotN5nbQrjEiNPTjS+9Pa98I+zb44q8xCRjMHZ1S36yo/LCsxlZBhGPJcT3W7Ltdy5VQcWauW3n/1fZ352ZneAzAkrN0/nkb9vY26OMTz2jtiCgAANkORAwAAAOijdgWL1uNBChth2ty5fkfzJ+eVPpDWzrmdmjk4o93zu2WtWKrbdTkVR3bRVmo61XilU0qmk5raM6VPr346sCJHrxtiB0lijlPik7+o/0q/vrcgMR11gSUuYwhiUM/ovTca1+62t8iglh8DAABbF0UOAAAAYMwVbhWU3JFUajql5I6kzKSpul2XtWLJtVz53trMkQlDZsKUmTRlpkwlUonGayqhpU+WlDmW6WkcUTaN7tQ2SpuNx9vtNdEu0TrMAkQckrKjikWU7y2IKBudD8oon61+GdQm7s1nLmyfWyGmAABgMChyAAAAAH1w6NAhFQqF9X9ns1lJ+tqxoMfz+by+973vSZJu3LihU6dOdXz/83d/LhmSYRryPV9O2VH5YVl1uy7f82WXbLmWK8/15Hu+5K8teWVKxkSjTf7jfM9Fjn5r9xffraImP0e1fFEcChytRpEw7kfSOkhMh31vnYprzfHE8RmQ4vuMjnNMAQDA4FHkAAAAAHr01ltvKZ/Pb7qcVNDlqzYe3/hZkPeNA5LneHJKjqqPq0pMJeRUHBmGIddyVVutySk7ci1XdafeKHjU/UbRo0/aJRk7LVHTqW3Udt2WxWm3GfMgN2mOe/J4mLHYeP3WY0G+86YwBY44xH1jUj6OhhGrTn2E+e6b4h5TAAAwHBQ5AAAAgD7I5XJ9O77xs9OnT3d//9ppffann8ku2aquVDW5PCnDMGQXbRkTjc3GnbKj2mpNdtmWU/l6scMwDeVObD6efgibvO61XZjrD0OcEu3txHlsmwkb006J8HH4fgZt1AWO5vFB/rwDAICtiyIHAADYsgzDWH/facPmdud1a9/p86B9A/2QXcjKqTiyVixVHlWUmEzIcz2lplMyE6Z8z5drubLLtmpParJLa4WOiivXarwyR+O1VNWo9DvZO84J9LiOPa7jGlfEEwAAjDtz1AMAAAAYhGaRoVlg2Fh0CHpe67JBre2an/u+/7V2nT4DBuXwK4dVzBdVWa6otFRSMV9svApFlZZKKi+XVXlUUfXLqmpPauuzOqwnlo68emTUww9lXJKxYZPH3ZaIiksyOuo4+zHuKDFt92o9Z9A6zSbpNaZRn5m4PFNRsVQVAACQKHIAAIAtrFmg6DaTIuh5g1T9oqq7//6u7v77u6o+ro5sHBhfi1cX5VSc9aJGMV9U8UGj0FFaKqn8eVmV5YqqX1RVfdwodFhPLMmTzlw5M9CxdUvAhm3X7Zygid2w7cLo9VqtYxxUMrfXWEQdZy/fW5wT8kGe9WHFNOjP17DjGfa/B/2IKQAA2NpYrgoAACCC1hkarRtCB12u6j/8z/9BH/8fH8su20pOJSVJbtVVcjqp4//9cf2D/+0fDGD02KrOf3Rel1++rOL9opyKo2Q6qUQqIZmSX/dVd+pyK+76DA7f93Xh5oW+9d8tqdpus+FuOm1SHfQaUfqNauO1g/z1fuuxTvcVtXDTqX2UWEQdZ9TvrZeYRhU1plGfrUHFtNt3HjaeUeMS9Lvv1zMKAAC2D4ocAAAAETSXpNrs350+a1r9m1X99PhPJU+amZ/RU/ueamwQ7Xiyy7aqj6r6xb/6hX75b3+pH3/8Y+2c2zmcG8NYy53I6dwH5/TOyXdkrVqa2jOlRCohY8KQ7/nyXE+u5a7P4Lhw84IyC4PfiyNIcjZMu27tB9VuWIY5vl762qrx70WnZ5qYdl5eK8p/J+J2fwAAYDQocgAAAAzZym9W9G/+3r9Ren9a+w/v13RmWskdSfmev755dHVvVVPLU1p9sKqfLvxU//gX/3jUw8aYmHtpTm/W3tT1H13X7fduKzGVkO+tFeBMQ67l6sirRwIvURU0MRtVr0nKqO0HmRztx7XDXmNU38Gw4j+oAs8g+hyHZ3qUP7NRrkExAwAAdEKRAwAAbFnNGRTdNv4Oel6//Ouj/1oz8zPKnchp9sVZTWenNZGakFt1Vf2yqspyRcmppMykKRnS6t+u6qfHfir9j0MZHraIs++e1dl3z2rpkyXlP85Lasz0yBwd/MwNAAAAABgWihwAAGBLahYtmoWLdstHdTuv9X1ru439tfa92Wd/9t/9mSYSE9p/eL/2f2e/Mkcz2j2/W3W3ruqjqpJLSRlmY2mhulOXa7lyLVcrv1mR/lzSf91zaLDNZI5llDlGYQMAAADA1kSRAwAAbFntNvxuPR70vDCft/vsl//2l5p7aU7T2WntenqX9vzWHs18a0bWl1ZjuaqqI7tsyy7ZSu1MKTWdUiqd0lMzT8n5fx2KHMAIhNn0mGV1AAAAgOEyRz0AAACA7eLh7YdKpBKa2j2l1HRqfa8Eu2ir7tRlyJA5YcpMmJpITmgiNaGJybXX1ISMCUNaHvVdAAAAAAAQH8zkAAAAW84zzzyjL774Yv3f+/btk6T1Y1NTU/rrv/5rffe735Uk/epXvxrK+8KtgmSqUazwJafqqLJcked48jxPdsmWa7vy6/76TBDDMGSYjeKHYRgy8sPZNwTAV5idAQAAAMQXMzkAAMCW8sd//Mc6efKkisXi+qup+e9arabPPvts/fiw3tsVW4YMea4nu2yr+mVVpaWSVvOrKn9eVvXLquxVW07FUb1WbxQ/6p58r1H0MGTItzsvoQUAAAAAwHbCTA4AALCl7N69W7t37970s29961vr7//wD/9w6O+zR7PyfV9O2ZG1YqnysCLDMJQqpmQmTNWdupyyo9pqTXbJllN1VLfq8mxPnuup7tdl5JjJAQAAAABAE0UOAACAIckdz8mxGgWO6hdVJaYS8uqeUtONIofv+XItV3bJbhQ6irbsii3HahQ75Eh+jpkcAAAAAAA0UeQAAAAYEjNp6rn/8jnlf5FvFDYmTHm2p+R0UhPJCfm+r7pdl1t1VSvW1gsdzdkdekESEzkAAAAAAFhHkQMAAGCIfvh//VD/Iv0vVH5YliS5lrte5JAhea4n13IbhY1iTbUna8WOii29OuLBAwAAAAAQM2w8DgAAMESJqYRe+dkrqn5RValQUjFfbLwKRZUKJZWXyio/LKvyqKLqF1VVV6pyLVc/fP+H/D83AAAAAABaMJMDAABgyL79yrdlTBi69uo11VZrmtw9qYnJCRmmIXlS3a3LtVzVVmtyqo5+eP2HOvQPD0m/GvXIAQAAAACIF4ocAAAAI/DimRf1zz//57ryB1e09Msl+b4vwzDky5c8yTANZRYy+kd/8Y80uWty1MMFAAAAACCWKHIAAACMyFN7ntKFv7yg1furevD/PNC9/3RPknTw1EHNvTSnXU/vGvEIAQAAAACIN4ocAAAAI7br6V3a9fQuffvst0c9FAAAAAAAxgrbVwIAAAAAAAAAgLFEkQMAAAAAAAAAAIwlihwAAAAAAAAAAGAsUeQAAAAAAAAAAABjiSIHAAAAAAAAAAAYSxQ5AAAAAAAAAADAWKLIAQAAAAAAAAAAxhJFDgAAAAAAAAAAMJYSox4AAAAAsF0YhrH+3vf90Od1ax+kXZD+AQAAAGBcMJMDAAAAGIJmoaFZXNis8NDtPN/32xYnDMNY/9z3/U3bdWoPAAAAAOOImRwAAADAkDQLDK1FiKjnbdamm2YxpJvCrYLyt/KSpNzxnLIL2U3PO/h2oG6/4d4b3dveeyPcNTder13bdn126yvofYYdc9B+gl53s/ZB2ra2G3Z/g+yz15i2u1bYZ6bXZyxK+0F8F/1+RqOOMcw1enneAABA/FHkAAAAALaIoMthdXJt8ZruXL+j5I6kZEhau4xTcXT4lcNavLrYh5EGc/Dt3hLfYc5pfjbKxGev42vXvlMcoxapuo213+OM2mc/v/OgsYoa0170O6aDiGec4rLxM4odAACMP4ocAAAAwBbRug9Ha6Gj0yyO+zfv69KpS0rPpjV/cl6p6ZQM05DneLJLtqwVS3c/vKuLkxd1/qPzyp3ISeqe0AySQOyUTA1T6Aii23jb9RckUdyPcUYdX7sZBkHj2PwsbCK69ZqDHudmfbZeN8j5YfuMol8x7SZqTKO26zWeUePSqe9O42nXbtDfPwAAGB725AAAAAC2ufs37+vKD64ou5DVs7//rOZemlPueE6Zoxnt//Z+7X1+r2aendG+F/YpfSCtyy9f1oO/ejDQMUVJZkZJjkfpb1D6Mb7W84LEJGpSPWwxqNN5QZcbande2ONB+gzTf+t1R/E8hY1plHa9xDMucel2HAAAjB+KHAAAAMCQNJeT6rbPRtDzNmsTxaVTl7T3ub3KLmSVO57T07/ztLLfy2r/d/Zrz3N7NHNwRrvmdmk6M62d2Z1KPpXUOyffidzfIIwqYTmKJXjajaFbMnfYY23tL67jDCqOSfGoMR337wIAAGAjlqsCAAAAhqC5iXizGNFuaalu57W+b7dJedClqq4tXlN6f1r7Xtin2RdnlVnIaObgjOp2XeWHZSWmEjIMQ57rqW7X5VqunIoja9XS+6++rzM/O9NTXHoVlyRsHBPgW8Wo904YdXGIZ+srxAYAAGyGmRwAAADAkPi+v/5qPR70vNZXkHab9dF05/od7Xluj9IH0to5t1MzB2e0e363dszu0NTMlCZ3TSo1nfrqlU4pmU5qas+UPr36aZQwBBJmI/F+JDrjUixpJ+7jG6aDb3/91ct1gp4z6g3px+H7H9XG4u367TYbJQ7fLQAA6A9mcgAAAADbVOFWQckdSaWmU0ruSMpMmqrbdVkrllzLle+tzSqZMGQmTJlJU2bKVCKVaLymElr6ZEmZY5mexhFl0+ign4c1qH0a+iUOCdl7b3yVXG638fggDCpZPexnLExf41DcaDWoeEWJzcZnNcg1AQDAeGImBwAAADBghULha6+mX/3qV+vvb9y4MfT3+Vt5yZAM05Dv+XLKjsoPyyrmi6o8qsgu2XItV57ryfd8yV9bJsuUjIlGm/zH+fAB6YN+Jn/j/hfdcR5fv2ZVhNHcwLqXjayDxHTYBYZuG7nHueAx6Gd0nGMDAAAGj5kcAAAAwAD95Cc/0fPPP6/Tp09/47ONS0iN6r18yXM8OSVH1cdVJaYSciqODMOQa7mqrdbklB25lqu6U28UPOp+o+jRJ+0So91mC/Rzmao4FhCk+I6v3VJAg96wul2yu92zspkwBY44xL15f3E1ylh1i02nsYV5ZgAAQLxR5AAAAAAG7Ne//rVee+21bxw/evTo+vuNRZBhvc8dz0mS7JKt6kpVk8uTMgxDdtGWMdHYbNwpO6qt1mSXbTmVrxc7DNNQ7kSu4733olvyOmpyM8w5g2gbpz56FeexbSZsTHt9xra6OMeg29jCFscAAEB8UeQAAAAAtqnsQlZOxZG1YqnyqKLEZEKe6yk1nZKZMOV7vlzLlV22VXtSk11aK3RUXLlW45U52tt+HKMS5+SsFP/xdRLXscd1XOOKeAIAgLigyAEAAABsY4dfOay7H95tFDYmGhuPp6ZTMpOm5Et1py6n4sguNQodzVkd1hNLR149MpIx9zo7o9fk7KCXDgo7vqAzXkadjI46zn4s1xQlpv26Vq863XuvMY36zMTlmYrzMl4AAGB42HgcAAAA2MYWry7KqTgqFooqLZVUzBdVfFBUMd/4d/nzsirLFVW/qKr6uKrak5qsJ5bkSWeunBno2AaRwOxncnYQCd5+F2AGlQRud92g4486zs3OG5eEfCfd4ikNL6bd2sWlyBM0NkHaAwCA8cZMDgAAAGCbO//ReV1++bKK94tyKo6S6aQSqYRkSn7dV92py6246zM4fN/XhZsX+tZ/t2Rjv5KpG/vp9tfxo9DL+DbOdtisbbcCQLfjYfdEaSfqOLu1a2cU33nUmEZNug8qpt2+87DxjNuz1qk/AAAwXpjJAQAAAGxzuRM5nfvgnEqPSlr5zUpjNke+qFKhtD6bo7xcVrFQlFNy9PqHryuzMPi9OO69Eb8EZJxnBXRKZg+zr279RR1nL33GXafxjyqmcdFLbDp9vhWeGwAA0MBMDgAAAACae2lOb9be1PUfXdft924rMZWQ7/mSJMM05Fqujrx6JPASVUGSh8NMvverv0EmRUcxvl76HEXbYd5f1GsO695G0e+4PS/9ag8AAOKNIgcAAACAdWffPauz757V0idLyn+cl9SY6ZE5OviZGwAAAAAQFkUOAAAAAN+QOZZR5hiFDQAAAADxRpEDAAAAsWIYxvp73/cjnRfkGoZhRGoHBBVms2SW0wEAAACiYeNxAAAAxEazyNAsMGwsOgQ9r1m8aL42u0a7Y93aAQAAAADihZkcAAAAiJVm4aJboaHded1mYDSLGf0oYhRuFZS/tbZvxfGcsgvZnq+JrYPZGQAAAMDgUeQAAADAltOu6LHZElVRXFu8pjvX7yi5IykZktYu6VQcHX7lsBavLvbcBwAAAACgO4ocAAAA2HI2K2x0K3AEmd1x/+Z9XTp1SenZtOZPzis1nZJhGvIcT3bJlrVi6e6Hd3Vx8qLOf3ReuRO5vt0TAAAAAOCbKHIAAABg22gtYrQWPlqLIxvdv3lfV35wRdmFrPa9sE/pA2kldyTle76ckqPqSlWVRxWlplMqFoq6/PJlnfvg3GBvCAAAAAC2OYocAAAAiJWge2a0O6/djI3WY51mdmz22aVTl5RdyCq7kNWXp5lJAAAgAElEQVTsi7PaObdTZtKUU3ZUfVzV5PKkEpMJmROmJKl4v6h3Tr4j/UnXWwYAAAAARESRAwAAALHRLFo0Cxft9tPodF7Yjcg3Xr9dm2uL15Ten9a+F/Zp9sVZZRYymjk4o7pdV/lhWYmphAzDkOd6qtt1uZYrp+LIWrVkX7MltugAAAAAgIEwRz0AAAAAYCPf99dfrceDnNftsyjXu3P9jvY8t0fpA2ntnNupmYMz2j2/Wztmd2hqZkqTuyaVmk599UqnlEwnNbVnSroT5u4BAAAAAGFQ5AAAAAA6KNwqKLkjqdR0SskdSZlJU3W7LmvFkmu58r21WSUThsyEKTNpykyZSqQSSqQSUkIyljovvQUAAAAAiIYiBwAAAGKhUCh87RX2+I0bN9Y/6+f7/K28ZEiGaTQ2GS87Kj8sq5gvqvKoIrtky7Vcea4n3/Mlf23pK1ONwodhSvk+BAgAAAAA8A3syQEAAICR+8lPfqLnn39ep0+f/sZn7ZacardfR+tnfXnvS57jySk1NhlPTCXkVBwZhiHXclVbrckpO3ItV3Wn3ih41P1G0QMAAAAAMDAUOQAAABALv/71r/Xaa69943gul9v0/Nbj2Wx2/f3GYkmv7wu7GrNH7JKt6kpVk8uTMgxDdtGWMdHYbNwpO6qt1mSXbTmVlmKH78vPUewAAAAAgEGgyAEAAAB0kF3Iyqk4slYsVR5VlJhMyHM9paZTMhOmfM+Xa7myy7ZqT2qyS2uFjorb2LPD9aXMqO8CAAAAALYmihwAAABAF4dfOay7H95tFDYmGhuPp6ZTMpOm5Et1py6n4sguNQodzVkd1hNLOjzq0QMAAADA1kWRAwAAAOhi8eqiLk5eVLFQlCS5lqtUOiUzZcowGktWuZYrp9JYtqr2pNYocHiSFiWxWhUAAAAADIQ56gEAAAAA4+D8R+fllBwV7xdVzBe1ml9VKV9SsVBUaamk8nJZleWKqo+rqjyuyPd9Xbh5YdTDBgAAAIAtjSIHAAAAEEDuRE7nPjin0qOSVn6zomK+UewoFUqNIsfnZZWXyyoWinJKjl7/8HVlFtiMAwAAAAAGieWqAAAAgIDmXprTm7U3df1H13X7vdtKTCXke421qAzTkGu5OvLqEZ25cmbEIwUAAACA7YEiBwAAABDS2XfP6uy7Z7X0yZLyH+clNWZ6ZI4ycwMAAAAAhokiBwAAABBR5lhGmWMUNgAAAABgVChyAAAAAENiGMb6e9/3I53X7RpBPu/UNwAAAACME4ocwBbVaxIlagIlaL8AAGw3zd+Rvu/LMIy2xYZO57W26fbvdtcGAAAAgK2CIgewBfUjibIxmdKvdgAAbHcbf192+l3Z7rxuBYwgnwf9HV24VVD+1tp+I8dzyi5kNz3v4NuBLvcN997o3vbeG+GuufF67dq26zNIX/0eb5g+gl57s/ZR7m3Y/Q2yz15j2u5a3dqHjWmvz9ewvot+P6OD+tkL+t+mfvzcAgCA0aLIAWxRvSZR+nX9bh7/9WP97f/9t7r3n+5Jkg6ePqhn/v4z2vvc3sjXBABgu+o00zLoDMtri9d05/odJXckJUPSWjOn4ujwK4e1eHWxn0Pu6ODbvSW+w5zT/GyUCc9ex9eufac4Ri1SdRtrv8cZtc9+fudBYxU1pr3od0wHEc9RxAUAAGwPFDkAjETxQVFXfnBFX/x/X0iSElMJ+b6vT9/9VIZhaPbQrM79xTmlD6RHPFIAAOJps8LFZoWNoAWO+zfv69KpS0rPpjV/cl6p6ZQM05DneLJLtqwVS3c/vKuLkxd1/qPzyp3ISeqe0AySQO6UTA1T6Aii23iD9DfIQkjU8bWbYRD0vpqfhU1Et15z0OPcrM/W6wY5P2yfUfQrpt1EjWnUdr3GM2pcOvUd9tw4FDUBAED/mKMeAIDt59PLn+rtv/u27KKtZ77/jH7r5d/St05/S/O/O6+nX3pa+57fp9UHq/qX8/9S//m9/zzq4QIAEDthNw9vLi/ZnOnROgvz/s37uvKDK8ouZPXs7z+ruZfmlDueU+ZoRvu/vV97n9+rmWdntO+FfUofSOvyy5f14K8e9PWeWoVJPgZNWAZJvI5SP8bXel6QmERNqodJgHc7L+hSTO3OC3s8SJ9h+m+97iiep7AxjdKul3iOKi4AAGDro8gBYKh+9X/+Sv/uf/h3eubvP6ODpw5q7u81kigHvntAs9+ebSRRvjWjvc/t1e5nduvPf/znun3l9qiHDQBAX7QrMoQ5L2yBw/f9r72axza6dOqS9j63V9mFrHLHc3r6d55W9ntZ7f/Ofu15bo9mDs5o19wuTWemtTO7U8mnknrn5DuBxzAM2zl5GrQAMOzlglr7i+s4g4rjMxY1puP+XfRiK94TAADbHctVAVtU0M1Fw25C2ku7WrGmP7vwZ3rm+8/owHcPaP939mtnbqfMhCm7bKvyqKLyw7ImUhOSKfmeL8/19P5/+770v4j/YgEAxlrz92bzd2e7PTO6nbfxfzd+HmaT8o2uLV5Ten9a+17Yp9kXZ5VZyGjm4Izqdl3lh2UlphIyDEOe66lu1+VarpyKI2vV0vuvvq8zPzsTJRx9E4f9D+KY/N5KRr200KiLQzxfX+lnbIgrAABbBylDYAvqZxJl4/te2109c1W753Zrz9/do32H9ilzNKOZZ2fkVByVPy9rIjkh+ZLneHItV261kUSprdRkvWdJ5/oUIAAARqRd4aHT3hpBjgf9fLNz7ly/o/mT80ofSGvn3E7NHJzR7vndslYs1e26nIoju2grNZ1qvNIpJdNJTe2Z0qdXPx1YkSPMRuL9SFb2ksgeRhKevz7/Sr8S3cN+xqKKwxiCGGXBMe6xAQAAg0WRA9iiRpVEaXe8Xqvrs//4meZ/b17pv5PW9IFp7cztVPrvpGWtWHKqjpLFpJLplteOpCZ3T8q6a0lexyEBAICQCrcKSu5IKjWdUnJHUmbSVN2uy1qx5FqufG/tDxsmDJkJU2bSlJkylUglGq+phJY+WVLmWKancUTZNDro52GF7W/Yid04JHPvvdG47802mB5kPNpdu9dE9yg3mu/W1zgWtwYVr37EhqIIAABbE3tyABiK/K28EpMJpdKpxrIXE4bsSmOJKqfsyHcbxRHDNGROmDITpiaSE5pINV5GwpCRD7ekFgAAcXHo0CEVCoX1V9PGYxuP37x5c/39jRs3BvY+fysvGY3fv77nyyk7Kj8sq5gvqvKoIrtky7Vcea4n3/Mlf22mpikZE402+Y/zkePSi34mf3vZuHzjsUElpOOcmG0WO5qvYWhuYN3LRtZBYhqHAtYwnq9+GPQzOs6xAQAAg0eRA9hCZmdn9f3vf1+fffbZ+kuSrl+/vn7OqN7/xaW/kDlhSoZUd+qyS7YqDyvrSZTaak1u1ZXnePLqnuSr8VpLvJimKS2FjQgAAKP31ltv6dSpU9/Y/FvafFPw5vFhvW8uFemUHFUfV1VaKqmYL6r8eVnVL6qqrdbklB25lqu6U28UPOp+o+jRJ61J643J682Sl4NYpiqua/vHtcDRrsDQS+EhaL/tjgVNdIcpcMQh7nEYQyejjFXcYwMAAIaD5aqALeSP/uiP9Pjx428cP3To0Mjfzz07p0f1R6rXGgWO6hdVTaQmZJdtGYYht+bKLtqyy42/GK3X6qq79fUkiu/7UipYHAAAiJtcLhfq+O/+7u+uvz99+vTA3ueON/q3S7aqK1VNLk/KMAzZRVvGRGOzcafsqLZak1225VS+XuwwTEO5E5vfQz90WhZJ6pzUHrdE9mbiPj4p3mPbTNiY9vqMbXXjFINxGisAAAiHIgewxezdu/cbx377t3975O9f+m9e0i8v/lJ2yZb1paXyw7KkRlLFnDAbSZTKWhKltCGJUqs3Eim+Jw0uhwIAwLaUXcjKqTiyVixVHlWUmEzIcz2lplMyE6Z8z5drubLLtmpPNvyOrrhyrcYrc7S3/ThGJe4Jz7iPr5O4jj2u4xpXxBMAAMQFRQ4AQ7H/O/slT7K+bCRRJiYnvkqiJL9KojhlR7UnNdVKjaUxnKojt+o2lq6aHfVdAACw9Rx+5bDufni38Tt5orHxePP3s/zGMpNOxZFdahQ6mrM6rCeWjrx6ZCRj7nV2Rr+Ts/3eDyDs+ILOeBl1MjrqOJvtehElpv26Vq863XuvMY36zMTlmQq7RBkAANia2JMDwHAY0rELx7SaX1V5udxY77tQVDHfeJWWSio/LKvyqKLql1VZK9b6rI5asSYdl3z1b+1vAADQsHh1UU7FUbFQXN+Po/hgw+/nz8uqLFdU/aKq6uOqak9qsp5YkieduXJmoGMbRGIyanK23Vg2Ho/DHiGt4xz0Zujtjncbf9RxRtmjJS4J+U768Xz1K6bd2sWlyBPlZy/OzwAAAIiOmRwAhuYP/vc/0O33bqv4oCj5kmu5SqVTmkhOSIbk1b3GbI6KI7vY+AtR60tLkzsn5f5Xa7M5AABA353/6Lwuv3xZxftFORVHyXRSiVRCMiW/7qvu1OVW3PUZHL7v68LNC33rv1tSdRAzLrr9dXy39oPQy/g2znbYrG3Ye2o9HnZPlHaijrNbu3Z6/c6jiBrTqM/XoGLa7TsPG89hP2sAAGD7YCYHgKH6o9t/JN/39eRvnjT+SrT5l6KF0tdnc3xRVWW5osRTCf2TT//JqIcNAMCWljuR07kPzqn0qKSV36x8NdOy+fv587LKy2UVC0U5JUevf/i6MguD34vj3hvx+cvrTuOIyzg7JbOH2Ve3/qKOs5c+467X52sQMY2LXmMzDjN5AABAb5jJAWCo0pm0/umn/1Q//d5PVbxfVOKphJI7kjImDPm+L89pbEBer9W1Y/8O/fjWj/XU3qdGPWwAALa8uZfm9GbtTV3/0XXdfu+2ElMJ+V5jGqVhGnItV0dePRJ4iaogCcVhJt/70d+gk6T9uH7Ya/TS5yjaDvP+ol5zWPc2in7H7XnptS0AABgPFDkADN2O/Tv0z/7mn+nGT27oF//qF3ry4ImSTyUlSW7F1VOzT+n7/9P39Xv/6++NeKQAAGw/Z989q7PvntXSJ0vKf5yX1JjpkTk6+JkbAAAAABAWRQ4AI3PqrVM69dYpOWVH+VtrSZTjOSV3JEc8MgAAkDmWUeYYhQ0AAAAA8UaRA8DIJdNJHfwvDo56GAAwVgzDWH/v+37o87q17/R50L6B7S7MZsksqQMAAABEw8bjAAAAY6ZZZGgWGDYWHYKe5/t+2wJFp3aGYay39X2/bd8AAAAAAAwDMzkAAADGULMA0a3QEPS8oO2izNwo3Cp8bVnC7EI29DWAccTsDAAAAGDwKHIAAABgIK4tXtOd63caey0ZktbqI07F0eFXDmvx6uJIxwcAAAAAGH8UOQAAABBJc+mqVvdv3telU5eUnk1r/uS8UtMpGaYhz/Fkl2xZK5bufnhXFycv6vxH55U7kRvB6AEAAAAAWwFFDgAAAITWqcBx5QdXlF3Iat8L+5Q+kFZyR1K+58spOaquVFV5VFFqOqVioajLL1/WuQ/OjeAOAAAAAABbAUUOAACAMdQsMnTbZyPoeWHatStwSNKlU5eUXcgqu5DV7Iuz2jm3U2bSlFN2VH1c1eTypBKTCZkTpiSpeL+od06+I/1JqOEBAAAAACCJIgcAAMDYaRYfmgWIjQWHjQWIbue1vg/TbrPNyK8tXlN6f1r7Xtin2RdnlVnIaObgjOp2XeWHZSWmEjIMQ57rqW7X5VqunIoja9WSfc2W2KIDAAAAABASRQ4AAIAx1G4mRevxoOdFvf5Gd67f0fzJeaUPpLVzbqdmDs5o9/xuWSuW6nZdTsWRXbSVmk41XumUkumkpvZMyb5jdxwPAAAAAACbMUc9AAAAAIy/wq2CkjuSSk2nlNyRlJk0VbfrslYsuZYr31ubHTJhyEyYMpOmzJSpRCqhRCohJSRjKdySWgAAAAAAUOQAAAAYI4cOHVKhUFh/SdKNGzfWPx/V+5+/+3PJkAzTaGwyXnZUflhWMV9U5VFFdsmWa7nyXE++50v+2pJXphqFD8OU8qHDAQAAAADY5liuCgAAYEy89dZbyufzHZekGtX7xgHJczw5pcYm44mphJyKI8Mw5Fquaqs1OWVHruWq7tQbBY+63yh6AAAAAAAQAUUOAACAMZLL5b5x7PTp06N//9ppffann8ku2aquVDW5PCnDMGQXbRkTjc3GnbKj2mpNdtmWU2kpdvi+/BzFDgAAAABAOBQ5AAAA0LPsQlZOxZG1YqnyqKLEZEKe6yk1nZKZMOV7vlzLlV22VXtSk11aK3RU3MaeHa4vZUZ9FwAAAACAcUORAwAAAH1x+JXDuvvh3UZhY6Kx8XhqOiUzaUq+VHfqciqO7FKj0NGc1WE9saTDox49AAAAAGAcUeQAAABAXyxeXdTFyYsqFoqSJNdylUqnZKZMGUZjySrXcuVUGstW1Z7UGgUOT9KiJFarAgAAAACEZI56AAAAANg6zn90Xk7JUfF+UcV8Uav5VZXyJRULRZWWSiovl1VZrqj6uKrK44p839eFmxdGPWwAAAAAwJiiyAEAAIC+yZ3I6dwH51R6VNLKb1ZUzDeKHaVCqVHk+Lys8nJZxUJRTsnR6x++rswCm3EAAAAAAKJhuSoAAAD01dxLc3qz9qau/+i6br93W4mphHyvsRaVYRpyLVdHXj2iM1fOjHikAAAAAIBxR5EDAAAAA3H23bM6++5ZLX2ypPzHeUmNmR6Zo8zcAAAAAAD0B0UOAAAADFTmWEaZYxQ2AAAAAAD9R5EDAAAAGBLDMNbf+74f6byonwEAAADAVsTG4wAAAMAQNAsQzeLDxoJE0PMMw5Dv++uvoJ8BAAAAwFbFTA4AAABgSJqFi25FiHbndZqdEWXmRuFWQflba/ulHM8pu5Dd9LyDb4e+tCTp3hvd2957o/1n7dp2atOpXdD27a4RpF0QUe+rU/so9zTs/gbZZ68xbXetsM/aoJ/NYX0X/X5Ge41Lp+v04+cdAACMN4ocAAAAwBjpx3JV1xav6c71O0ruSEqGpLVTnYqjw68c1uLVxb6OuZODb4dPXDY/G0Tyslu/vfbZ6321a99pbFGLVL3EIso4o/bZz2claKyixrQX/Y7pIOI5irgAAABQ5AAAAADGyMbiRXOJqiCfSdL9m/d16dQlpWfTmj85r9R0SoZpyHM82SVb1oqlux/e1cXJizr/0XnlTuQkdU9oBkkgd0qmdkrSRm0XdFxB2wbtM8q1g/bRboZB2HiETUSHjUWv49ysz9brBjk/bJ9R9Cum3USNadR2vcYzbFyCFGmiPDMAAGB7YE8OAAAAYBu4f/O+rvzgirILWT37+89q7qU55Y7nlDma0f5v79fe5/dq5tkZ7Xthn9IH0rr88mU9+KsHAx1TtyRpmMJHP3RKpvarz34UZVrPC5L8jZpU7yUWYcfZLZkd5XkIc99hi3ajSKqHjWmUdr3Ec1RxAQAA2xtFDgAAAGBImstJddsUvN15ndp1u+alU5e097m9yi5klTue09O/87Sy38tq/3f2a89zezRzcEa75nZpOjOtndmdSj6V1Dsn3wlyW9vOKJcq6pboH/bYWvuL6ziDimOCPmpMx/27kOI9NgAAEB8sVwUAAAAMQXMT8WYxot3SUp3O67QReafPri1eU3p/Wvte2KfZF2eVWcho5uCM6nZd5YdlJaYSMgxDnuupbtflWq6ciiNr1dL7r76vMz870+doDEfUjbYxfIPcZyVM/6Pqj2ezsyibqxNTAAC2D4ocAAAAwJC02wy89XinTcOjfHbn+h3Nn5xX+kBaO+d2aubgjHbP75a1Yqlu1+VUHNlFW6npVOOVTimZTmpqz5Q+vfrpwIocvW6I3c8lmOKAv1r/Sr8S1kFiGofnIg5jCGKcntFxiSkAAOgdRQ4AAABgCyvcKii5I6nUdErJHUmZSVN1uy5rxZJrufK9tZkjE4bMhCkzacpMmUqkEo3XVEJLnywpcyzT0ziibBrdqW3YTYiDJGfvvdE4b7NNlYeZ3I1DUnZUsWh37V4T1nHatHqUz1a/DCNeYfdI2awtAADYHtiTAwAAABiwQqHwtVeY43/5l3+5fvzGjRuh3+dv5SVDMkxDvufLKTsqPyyrmC+q8qgiu2TLtVx5riff8yV/bX8PUzImGm3yH+f7HpNehS2abDwW9K/7N74GLc5/dT7sWEhfbWDdy0bWQWI67GR4P57NUYnrMzrOMQUAAP3BTA4AAABggH7yk5/o+eef1+nTp7/xWdDlqzY7Huq9L3mOJ6fkqPq4qsRUQk7FkWEYci1XtdWanLIj13JVd+qNgkfdbxQ9+qRdYrTdbIFubYO02+w63RKe7TZjHuQmzXFPHg8zFhuv33oszHcepsARh7gHeTZHKU6xCiruMQUAAP1DkQMAAAAYsF//+td67bXXvnE8l8ttev7G4xvfbyyUBH1f2NWYIWKXbFVXqppcnpRhGLKLtoyJxmbjTtlRbbUmu2zLqXy92GGYhnInNh9nP4RNXvfaLsz1h2EcksdxHttmwsa0UyJ8HL6fQRtFDIg7AAAIgyIHAAAAsIVlF7JyKo6sFUuVRxUlJhPyXE+p6ZTMhCnf8+VaruyyrdqTmuzSWqGj4sq1Gq/M0d7249gq+p14HedEblzHHtdxjSviCQAAxgF7cgAAAABb3OFXDjf24FiuqLRUUjFfbLwKRZWWSiovl1V5VFH1y6pqT2rrszqsJ5aOvHpk1MPvmzgtXRM2edxtiai4JKOjjrMf444S03av1nMGLcgeM1FjGvWZGdUz1a+f0zj9vAMAgMGiyAEAAABscYtXF+VUnPWiRjFfVPFBo9BRWiqp/HlZleWKql9UVX3cKHRYTyzJk85cOTPQsXVLwPar3cbjUTai7mfCt9drtY5xUMncXmMRdZybnRfXhHwYvT6bm10jakyD/nyNMp5h9l7pdDzOzwQAAOgPlqsCAAAAtoHzH53X5Zcvq3i/KKfiKJlOKpFKSKbk133Vnbrcirs+g8P3fV24eaFv/Qfd8DtMm17adTLIvwDfeO0gf73feqzZZrO23QoA3Y73K5ZRx9mtXTu9xDSqqDGN+mwNKqbdvvOw8ezlWYuKGRsAAICZHAAAAMA2kDuR07kPzqn0qKSV36ysL1lVKpTWZ3OUl8sqFopySo5e//B1ZRYGvxdHu+WAuiVBo7QLsvRQp2RxHP4ivNP4htlXL3EcVJ9xN8hncxDtRiHqMm7tPovjPQIAgP5jJgcAAACwTcy9NKc3a2/q+o+u6/Z7t5WYSsj3fEmSYRpyLVdHXj0SeImqIAnEXpKMUdv2mtgcZGK0H9cOe41RfAe9tB3m/UW95rg9m2Hajep5idqeQgYAAKDIAQAAAGwzZ989q7PvntXSJ0vKf5yX1JjpkTk6+JkbAAAAANBPFDkAAACAbSpzLKPMMQobAAAAAMYXRQ4AAADEimEY6+993w99Xrf2Qa5vGEbHvoGmMJses6wOAAAA0H9sPA4AAIDYaBYgmgWGjQWJoOf5vt+1eNF8bXb9dn0CAAAAAOKHmRwAAACIlWaBol0RIux5m7Vpp1kECXq9wq2C8rfW9rQ4nlN2IRuoHbYOZmcAAAAAo0WRAwAAAFC4JaquLV7Tnet3lNyRlAxJa82ciqPDrxzW4tXFwQ0UAAAAALCOIgcAAAC2pY1FjaAFjvs37+vSqUtKz6Y1f3JeqemUDNOQ53iyS7asFUt3P7yri5MXdf6j88qdyA36NgAAAABgW6PIAQAAgG1ns6JG6xJVrefcv3lfV35wRdmFrPa9sE/pA2kldyTle76ckqPqSlWVRxWlplMqFoq6/PJlnfvg3FDuBwAAAAC2K4ocAAAAiJWg+2KE3T+jtd1GmxU8Wo9dOnVJ2YWssgtZzb44q51zO2UmTTllR9XHVU0uTyoxmZA5YUqSiveLeufkO9KfhBoeAAAAACAEihwAAACIjWbRolm42Fho2Fh46HZe6/uNy1K1nhNkmapri9eU3p/Wvhf2afbFWWUWMpo5OKO6XVf5YVmJqYQMw5DneqrbdbmWK6fiyFq1ZF+zJbboAAAAAICBoMgBAACAWGlXdOg2+6Lb8W6fdTrvzvU7mj85r/SBtHbO7dTMwRntnt8ta8VS3a7LqTiyi7ZS06nGK51SMp3U1J4p2XfsQH0CAAAAAMIzRz0AAAAAIM4KtwpK7kgqNZ1SckdSZtJU3a7LWrHkWq58b21WyYQhM2HKTJoyU6YSqYQSqYSUkIylcEtqAQAAAACCocgBAACAWDh06JAKhcL6q+nGjRsjfZ+/lZcMyTCNxibjZUflh2UV80VVHlVkl2y5livP9eR7vuSvLYdlqlH4MEwpHz4eAAAAAIDuWK4KAAAAI/fWW28pn89vupzUxmOjei9f8hxPTqmxyXhiKiGn4sgwDLmWq9pqTU7ZkWu5qjv1RsGj7jeKHgAAAACAgaHIAQAAgFjI5XKbHj99+vRI3+eON8Zll2xVV6qaXJ6UYRiyi7aMicZm407ZUW21Jrtsy6m0FDt8X36OYgcAAAAADAJFDgAAAKCD7EJWTsWRtWKp8qiixGRCnuspNZ2SmTDle75cy5VdtlV7UpNdWit0VNzGnh2uL2VGfRcAAAAAsDVR5AAAAAC6OPzKYd398G6jsDHR2Hg8NZ2SmTQlX6o7dTkVR3apUehozuqwnljS4VGPHgAAAAC2LoocAAAAQBeLVxd1cfKiioWiJMm1XKXSKZkpU4bRWLLKtVw5lcayVbUntUaBw5O0KInVqgAAAABgIMxRDwAAAAAYB+c/Oi+n5Kh4v6hivqjV/KpK+ZKKhaJKSyWVl8uqLFdUfVxV5XFFvu/rws0Lox42AAAAAGxpFDkAAACAAHIncjr3wTmVHpW08psVFfONYkepUGoUOT4vq7xcVrFQlFNy9PqHryuzwGYcAAAAADBILC9teBgAACAASURBVFcFAAAABDT30pzerL2p6z+6rtvv3VZiKiHfa6xFZZiGXMvVkVeP6MyVMyMeKQAAAABsDxQ5AAAAgJDOvntWZ989q6VPlpT/OC+pMdMjc5SZGwAAAAAwTBQ5AAAAgIgyxzLKHKOwAQAAAACjQpEDAAAAGBLDMNbf+74f6bxBfAYAAAAA44qNxwEAAIAhaBYZmgWGjUWHoOcZhiHf99dfrZ9FaQcAAAAA44yZHAAAAMCQNAsQ3QoN7c7rNgMjarvNFG4VlL+1tt/I8ZyyC9lNzzv4duhLS5LuvdG97b032n/Wrm2nNkGuEaR9a7swfYa9dtg+hn1P/epvkH3241nZ7Frd2oeNaS8/D+3aD+K76Pcz2mtc2l0najsAADB+KHIAAAAAY2TQy05dW7ymO9fvKLkjKRmS1rpwKo4Ov3JYi1cX+95nOwff3jwB2Sl52fwsSpK2U5/d2vXDoO5rEPcUNYad2kZt16ltrzENeq0o5/VTv2M6iHiOIi4AAGB7oMgBAAAAjJGNhY3mMlRhtGtz/+Z9XTp1SenZtOZPzis1nZJhGvIcT3bJlrVi6e6Hd3Vx8qLOf3ReuRM5Sd0TmkESyJ2SqZ2StFHbBWkfpN0gkrbd4hkk+bzx86DxiHpP7f56flDj3KzP1usGOT9sn1H0K6bdRI1p1Ha9xjNsXIIUacKMs1s7AAAwftiTAwAAANgmOhU4rvzgirILWT37+89q7qU55Y7nlDma0f5v79fe5/dq5tkZ7Xthn9IH0rr88mU9+KsHAx1rtyRpmMLHRt2Sm1H77YcgieGw1whSMIiaVA+bWO503qC+t37ENEj/rdcdRfI8bEyjtOslnqOKCwAA2PoocgAAAABD0lxqqtvG3+3Oi9queazdrI9Lpy5p73N7lV3IKnc8p6d/52llv5fV/u/s157n9mjm4Ix2ze3SdGZaO7M7lXwqqXdOvtNxLBieoAWAYS8X1NpfXMcZVBwT9FFjOu7fhTSYpdYAAMB4YrkqAAAAYAiam4E3CxDtlp3qdF6nDcW7XX/j/278/NriNaX3p7XvhX2afXFWmYWMZg7OqG7XVX5YVmIqIcMw5Lme6nZdruXKqTiyVi29/+r7OvOzM/0J0BCwRM14GvX3NuriEM9rZ1HjQ1wBANg6KHIAAAAAQ9JuJkXr8U77bET5rFObO9fvaP7kvNIH0to5t1MzB2e0e363rBVLdbsup+LILtpKTacar3RKyXRSU3um9OnVTwdW5Oj1r7SDJDDHKZnMX59/pV/fW5CYjrrAEpcxBMEzCgAARoUiBwAAALBNFW4VlNyRVGo6peSOpMykqbpdl7ViybVc+d7a7JAJQ2bClJk0ZaZMJVKJxmsqoaVPlpQ5lulpHFE2je7UNkqbjcfjnEyOw9juvdGI1WYbTA8y0T2o7y3KRueDMsx4Dsow4hX1Ox+Hn3EAABAee3IAAAAAA3bo0CEVCoX1V9PGY63Hl5eXdePGjfVjg3j/83d/LhmSYRryPV9O2VH5YVnFfFGVRxXZJVuu5cpzPfmeL/lrS16ZkjHRaJP/ON9DZAYjaGK4uRFy3DdEjnNitlnsaL6GoR/fW5CYDrvA0G0j9zgXPOL8jAIAgK2PmRwAAADAAL311lvK5/ObLhnVbXmp1j03+v2+cUDyHE9OyVH1cVWJqYSciiPDMORarmqrNTllR67lqu7UGwWPut8oevRJu8Rou9kC3dpGbddphsKoxDV53G5j6kFvWN2P7y1MgSMOcW/eX1zFKVYAAGB7osgBAAAADFgul4t0/PTp0+vHBvL+tdP67E8/k12yVV2panJ5UoZhyC7aMiYam407ZUe11Zrssi2n8vVih2Eayp3Y/B76IWrRIY7FiijGIXkc57FtJmxMOxUXxuH7GbRRxIClqgAAQCuKHAAAAMA2lV3Iyqk4slYsVR5VlJhMyHM9paZTMhOmfM+Xa7myy7ZqT2qyS2uFjoor12q8Mkd7248DmxvnhGxcxx7XcY0r4gkAAOKCPTkAAACAbezwK4cbe3AsV1RaKqmYLzZehaJKSyWVl8uqPKqo+mVVtSe19Vkd1hNLR149MurhhzIuydiwyeNuS0TFJRkddZz9GHeUmLZ7tZ4zaJ1mk/Qa06jPzKieqajLdsV5uS8AANA7ihwAAADANrZ4dVFOxVkvahTzRRUfNAodpaWSyp+XVVmuqPpFVdXHjUKH9cSSPOnMlTMDHVu3BGzYdt3OiUMxoNcxtN7XoJK7vRZUoo4zyvcWh++1myDP+rBiGvTna5TxjNp3nJ8BAAAQHctVAQAAANvc+Y/O6/LLl1W8X5RTcZRMJ5VIJSRT8uu+6k5dbsVdn8Hh+74u3LzQt/67JVXbbS7eTadNqoNeI0if7TbfDmvjdYL89X7rsU731a0A0O141O9gs3FEGWfU762XmEYVNaZRC1KDimm37zxsPAf98wMAALYvZnIAAAAA21zuRE7nPjin0qOSVn6zsr5kValQWp/NUV4uq1goyik5ev3D15VZGPxeHO2WA+qWBO22jFCnpO84J1g73dcw+wry/YQ53o8+467b8zqKmMYNG44DAIB2mMkBAAAAQHMvzenN2pu6/qPruv3ebSWmEvI9X5JkmIZcy9WRV48EXqIqSEKxl6RjrwnLKO0HnSTtx/XDXmNU38GwlhsaZoGn1z5H8UyHbTduP7P96BcAAMQfRQ4AAAAA686+e1Zn3z2rpU+WlP84L6kx0yNzdPAzNwAAAAAgLIocAAAAAL4hcyyjzDEKGwAAAADijSIHAAAAAAxAmI2kWVIHAAAAiIaNxwEAAAAAAAAAwFhiJgcAAAAADACzMwAAAIDBYyYHAAAAAAAAAAAYSxQ5AAAAAAAAAADAWKLIAQAAAAAAAAAAxhJFDgAAAAAAAAAAMJYocgAAAAAAAAAAgLFEkQMAAAAAAAAAAIwlihwAAAAAAAAAAGAsUeQAAAAAAAAAAABjiSIHAAAAAAAAAAAYSxQ5AAAAAAAAAADAWKLIAQAAAAAAAAAAxhJFDgAAAAAAAAAAMJYocgAAAAAAAAAAgLFEkQMAAAAAAAAAAIwlihwAAAAAAAAAAGAsUeQAAAAAAAAAAABjiSIHAAAAAAAAAAAYSxQ5AAAAAAAAAADAWEqMegAAAADYnGEY6+993+/pPMMwvvFZu3ZB+wUAAAAAYNSYyQEAABBDzUJDs8iwsfAQ9rx2x3zfX3+1FjYobgAAAAAAxgEzOQAAAGKqWWhoLUKEOa9ZzGg93u8iRuFWQflbeUlS7nhO2YXspucdfDva9e+90b3tvTfCXXPj9bq13azvoP310jbKtcNcP+rYWtsNu79B9tlrTNtdK+wzFuWZ7LX9IL6Lfj+jYb6HQf7sAQAAID4ocgAAAGxRmy1R1fp5U9Six7XFa7pz/Y6SO5KSIWntMk7F0eFXDmvx6mKk60Zx8O3eEt9hz+3WX6c+mp/1knDt9fpR7itqkarbWPs9zqh99vM7CxqrqDHtRb9jOoh49hKXQf/sAQAAIF4ocgAAAGxB3Qoc0jf34QhT6Lh/874unbqk9Gxa8yfnlZpOyfj/2bvfGDfuPM/vn6om2W2xJbX+jUi2T4IwlsczWl2sHsk7u3eCNMlgjFtgcIjUBjRyAOPkfXBPDkIOSYBcYMw6UZAHAe5g3AJzQA5WHmilGQky8uCAdSIEa80pumTWWi9gn7LnHcXWbaupkWRtt/ini1VFVh5QbFM0/1QVi2RV6/0CCFHF+tXvV18Wn3y//fv9TEMNpyG7bMtasXTn2h2dmz6nMzfOqHCkIGlwQtNP4rFfMjVIocOPXn+JP6i/QX/BH9V4B8Uz6Pj8jqv1WdBEdOc1Rz3Obn12XtfP+UH7DCOqmA4S9TM9qN2w8Qwal3H99gAAABAf7MkBAACwQRmGsf5q/T8KSzeXdPEnF5VfyGvfj/Zp/rV5FQ4XlHs1p13f3aXt+7drbt+cdry8Q9ndWV14/YLu/fpeJH33EmYJmyBtOs/127bXeVEkV/0khoNew0/BIGxSPUgCfNB5fpdiChr/KGLqp//O604i2R7VMx02ZqN41vxcn8IGAADAxkORAwAAIKb8Fie6nde+qXj7nh2dbcI4f+y8tr+0XfmFvAqHC3rxBy8q//28dn1vl7a9tE1ze+e0ZX6LZnOz2pzfrPQLab1/9P3Q/Y1CkL0HBiVLJ7Hc0DDiel+d/cV1nH7FMZkeNqZJ/y4AAACwsbFcFQAAQAy1NgtvFSN6LS3V7zw/12//f/v1O9+3Pr+yeEXZXVnteHmHdr6yU7mFnOb2zqlu11V5UFFqJiXDMNRwG6rbdbmWK6fqyHpi6YNTH+jEL06ECUdkSMJufJPec2HSxaE4FlcAAACAUWImBwAAQEx1zsRoP+7nvH5tBl2/20wQSbp99ba2vbRN2d1ZbZ7frLm9c9q6Z6s27dykmbkZTW+ZVmY28/Urm1E6m9bMthl9evnTMGHwxU9ieZzJ70F/2T7qsVDM+dre9559DXMdv+dMstAw7H2Oy6jGOOnfHgAAAMaPmRwAAADwpXirqPSmtDKzGaU3pWWmTdXtuqwVS67lyms8nVUyZchMmTLTpsyMqVQm1XzNpHT/k/vKHcoNNY4wm0b7/TxKd8/2TziPs9gySe1x6LXx+CiMKskdt2esXRKKG51GtYn7pH97AAAAGB9mcgAAAMRMsVh85jXM8cePH+v69evrx4Z5/9GljyRDMkxDXsOTU3FUeVBRabmk6qOq7LIt13LVcBvyGp7kPV3uypSMqWab5Y+Xhw9QCElM/oYV579Uj2pWRRCtDayH2cjaT0zH/YwN2sg9zs98nJ9RAAAAJA8zOQAAAGLk3Xff1f79+3X8+PFvfNZrOapBx9s/H+Z984DUcBpyyo7WHq8pNZOSU3VkGIZcy1XtSU1OxZFruao79WbBo+41ix4R6ZUYHTRbYNwJ1X799hrrKPucpF5LCI16w+pehYAg8Q9S4IhD3Fv3F1fjiNUkfnsAAACYHIocAAAAMfP555/r9OnT3zheKBS6nj/oeHvBZKj3p4/ri59/Ibtsa21lTdMPp2UYhuySLWOqudm4U3FUe1KTXbHlVJ8tdhimocKR7mONwqDkdb/Eb9SJ10HXC5poj6LPOIjz2LoJGtNxPmNJNOkCR+s4hQ4AAICNhSIHAAAAfMkv5OVUHVkrlqqPqkpNp9RwG8rMZmSmTHkNT67lyq7Yqq3WZJefFjqqrlyr+cq9Otx+HOguyQn0uI49ruNKKuIJAACAUaHIAQAAAN8OvHFAd67daRY2ppobj2dmMzLTpuRJdacup+rILjcLHa1ZHdaqpYOnDk5kzGGXGfI7M2TSSdug40jKfYUdZxTLNYWJaVTXGla/ex82pmGfmbg8UwAAANiY2HgcAAAAvi1eXpRTdVQqllS+X1ZpuaTSvZJKy83/V35bUfVhVWtfrWnt8ZpqqzVZq5bUkE5cPDHSsY1yH4LOa/vtq9d5UY112ORx2Psatp/O44PGH2X8N0JC3s9zNa6YDmo36T1x/B4HAABAcjGTAwAAAIGcuXFGF16/oNJSSU7VUTqbViqTkkzJq3uqO3W5VXd9BofneXr75tuR9T8oSRllMrV9VkC3fget+9+r3aD2frRfN2gfYe/Lb+I46J4ovYw6/p2GiWlYYWMaNlk/qpgO+s6DxjNsXMbx2wMAAEC8MJMDAAAAgRSOFPTmh2+q/KislS9XmrM5lksqF8vrszkqDysqFUtyyo7euvaWcguj34vj7tnRJC77JX3DjmdUYw0i7H1F3ZefOAY5HkWfcddv/JOKaZzE/bcHAACAaDGTAwAAAIHNvzavd2rv6OpPr+qzX36m1ExKXsOTJBmmIddydfDUQd9LVPlJOo4z+R5l36NKqEZx3aDXmFQcwrYd5/2Fvea47m0S/U76d0MxAwAA4PlAkQMAAAChnbx0UicvndT9T+5r+eNlSc2ZHrlXRz9zAwAAAAAAihwAAAAYWu5QTrlDFDYAAAAAAONFkQMAACSaYRjr7z3PC3xev/btn/k5p1//wCBBNpJmGR4AAAAAaGLjcQAAkFitAkOruNCtKDHoPM/zehYnWp/1OscwjL6fAwAAAACA0WImBwAASLRWccHzvJ5FjiDn9dIqaPT6vx/FW0Ut33q6b8XhgvIL+cDjwMbF7AwAAAAACI4iBwAAQEh+l6q6snhFt6/eVnpTWjIkPT3VqTo68MYBLV5eHPFIAQAAAADYmChyAAAADNBr1sagmR1LN5d0/th5ZXdmtefoHmVmMzJMQw2nIbtsy1qxdOfaHZ2bPqczN86ocKQw8nsBAAAAAGAjocgBAAAwAks3l3TxJxeVX8hrx8s7lN2dVXpTWl7Dk1N2tLaypuqjqjKzGZWKJV14/YLe/PDNSQ8bAAAAAIBEocgBAAASrTWDYtA+G37Pi8r5Y+eVX8grv5DXzld2avP8ZplpU07F0drjNU0/nFZqOiVzypQklZZKev/o+9I/G8vwAAAAAADYEChyAACAxGoVLVqFi17LRw06r/O9nw3GOwsm7edcWbyi7K6sdry8Qztf2ancQk5ze+dUt+uqPKgoNZOSYRhquA3V7bpcy5VTdWQ9sWRfsSW26AAAAAAAwBeKHAAAINF6bfjdedzveUE+7/XZ7au3tefoHmV3Z7V5frPm9s5p656tslYs1e26nKoju2QrM5tpvrIZpbNpzWybkX3b7jseAAAAAADwNXPSAwAAANhIireKSm9KKzObUXpTWmbaVN2uy1qx5FquvMbTWSVThsyUKTNtysyYSmVSSmVSUkoy7o9nSS0AAAAAAJKOIgcAAEikYrH4zKvz+I0bN9aPXb9+fWzvP7r0kWRIhmk0NxmvOKo8qKi0XFL1UVV22ZZruWq4DXkNT/KeLpNlqln4MExpOUxEAAAAAAB4/rBcFQAASJx3331X+/fv1/Hjx7/xWfs+HJ3HxvG+eUBqOA055eYm46mZlJyqI8Mw5Fquak9qciqOXMtV3ak3Cx51r1n0AAAAAAAAvlHkAAAAifT555/r9OnT3zheKBSe+VfSM8WQkb8/fVxf/PwL2WVbaytrmn44LcMwZJdsGVPNzcadiqPak5rsii2n2lHs8Dx5BYodAAAAAAD4QZEDAAAgQvmFvJyqI2vFUvVRVanplBpuQ5nZjMyUKa/hybVc2RVbtdWa7PLTQkfVbe7Z4XpSbtJ3AQAAAABAMlDkAAAAiNiBNw7ozrU7zcLGVHPj8cxsRmbalDyp7tTlVB3Z5WahozWrw1q1pAOTHj0AAAAAAMlBkQMAACBii5cXdW76nErFkiTJtVxlshmZGVOG0VyyyrVcOdXmslW11VqzwNGQtCiJ1aoAAAAAAPDFnPQAAAAANqIzN87IKTsqLZVUWi7pyfITlZfLKhVLKt8vq/KwourDqtYer6n6uCrP8/T2zbcnPWwAAAAAABKFIgcAAMAIFI4U9OaHb6r8qKyVL1dUWm4WO8rFcrPI8duKKg8rKhVLcsqO3rr2lnILbMYBAAAAAEAQLFcFAAAwIvOvzeud2ju6+tOr+uyXnyk1k5LXaK5FZZiGXMvVwVMHdeLiiQmPFAAAAACAZKLIAQAAMGInL53UyUsndf+T+1r+eFlSc6ZH7lVmbgAAAAAAMAyKHAAAAGOSO5RT7hCFDQAAAAAAokKRAwAAIKYMw1h/73neUOcZhvHMZ+1tOtv2+wwAAAAAgDhh43EAAIAYahUa+hUe/J7Xq2jR/vL7GQAAAAAAccJMDgAAgJhqFRg8z+tZ5Bh0XmsGR7/2nbM8/H7WrnirqOVbT/cbOVxQfiHf9by97w28VFd3zw5ue/dssGu2X29Q2259++1vmLZhrh3k+mHH1tlu3P2Nss9hY9rrWkGfsTDP5LDtR/FdRP2MBvkeRvnbAwAAQHxQ5AAAANig/BYohnFl8YpuX72t9Ka0ZEh62p1TdXTgjQNavLw40v7b7X1vuMR30HMH9devjyBjDXt9Kdz4+o0tbJFqmFiMM/7DxtTvtcKcF6WoYzqKeA4Tl1H/9gAAABAvFDkAAAA2IL8FjrCzOJZuLun8sfPK7sxqz9E9ysxmZJiGGk5DdtmWtWLpzrU7Ojd9TmdunFHhSEHS4ISmn+Rjv2Rq1AnMXn+JH6S/zs+jHOugePpJPoe5r9ZnQRPRQWMxivh3XtfP+UH7DCOqmA4SNqZh2w0bz7Bx6db3qL9DAAAATAZ7cgAAAGxQhmGsv1r/j8LSzSVd/MlF5Rfy2vejfZp/bV6FwwXlXs1p13d3afv+7ZrbN6cdL+9QdndWF16/oHu/vhdJ372EWcImSJvOc/0uBRQk6RuUn8Rw0Gv4KRiETaoPE4so4x/muJ8+g/Tfed1JJNyDxjRMu2HiGTYu4/jtAQAAIF4ocgAAAMSU3+JEt/O6bR4e1dJV54+d1/aXtiu/kFfhcEEv/uBF5b+f167v7dK2l7Zpbu+ctsxv0WxuVpvzm5V+Ia33j74fSd9RCbL3wKCE+KiW1RmVcdxXGJ39xXWcfsUxoR42pkn/LrpJ0lgBAADQH8tVAQAAxFBrs/BW4aK9QNG+jFS/8wYJs1TVlcUryu7KasfLO7TzlZ3KLeQ0t3dOdbuuyoOKUjMpGYahhttQ3a7LtVw5VUfWE0sfnPpAJ35xwvf4RoHE5sYXZqbOKPqfVH9xLK4AAAAAo0SRAwAAIKZ6FSA6j/spbHQ7p1+7Xp/dvnpbe47uUXZ3VpvnN2tu75y27tkqa8VS3a7LqTqyS7Yys5nmK5tROpvWzLYZfXr505EVOfwklied/B4nijlfi6oIkJRnLA5j8INnFAAAAFGhyAEAAABfireKSm9KKzObUXpTWmbaVN2uy1qx5FquvMbTWSVThsyUKTNtysyYSmVSzddMSvc/ua/codxQ4wizabTfz6Ny92xznN02OB5ncjcOie5JxaLXtYctAsTlGevWVxILB1HHKy6/PQAAAIwPRQ4AAICY+c53vqNisbj+/3w+L0nPHAt6PJVK6fbt2zp27Jgk6fr164Hfz/6HWcmQDNOQ1/DkVBxVHlRUt+vyGp7ssi3XctVwG/IanuQ93SfElIypZpvlj5eHLnKEMcnk5qSWL4pDgaPTJL6HKBLdfmI67nvrtbF2axzdkvxxMa5nlKIGAADA84EiBwAAQIz87Gc/0/LycqDlpcIsaxX6vSc1nIacsqO1x2tKzaTkVB0ZhiHXclV7UpNTceRarupOvVnwqHvNokdEeiVGB/319rgTvr02Yx7lJs1xLXBMIhbt1+881utZ6SZIgSMOcW8vdMTROGI1qecNAAAAk0GRAwAAIGYKhcJIjh8/fnyo98UtzZkhdtnW2sqaph9OyzAM2SVbxlRzs3Gn4qj2pCa7YsupPlvsMExDhSPdxxqFQcnrfonNUSZex5X4jlOivZc4j62boDGd1DOWFOOOwfMcawAAgOcJRQ4AAAD4kl/Iy6k6slYsVR9VlZpOqeE2lJnNyEyZ8hqeXMuVXbFVW63JLj8tdFRduVbzlXt1/EtVxVHUyd4kJ9DjOva4jiup4hLPuIwDAAAA0aHIAQAAAN8OvHFAd67daRY2ppobj2dmMzLTpuRJdacup+rILjcLHa1ZHdaqpYOnDk5kzGGXGfI7M2TSydKg40jKfYUdZxTLNYWJaVTXGla/ex82pmGfmbg8UwAAANiYzEkPAAAAAMmxeHlRTtVRqVhS+X5ZpeWSSvdKKi03/1/5bUXVh1WtfbWmtcdrqq3WZK1aUkM6cfHESMc2ynX2O6/tp69e50SZ8B32WmHuK4p+Oo8PGn/YcXY7byMk5AfFUxpfTAe1m0Q8x/HbAwAAQHwwkwMAAACBnLlxRhdev6DSUklO1VE6m1Yqk5JMyat7qjt1uVV3fQaH53l6++bbkfU/KKkaZQKzfVZAt36DJpKj1H7tQX+93+1YmPvyk1zv1T5MLMKOc1C7XoaJaVhhYxr22RpVTAd950HjOcyzNqg/AAAAbCzM5AAAAEAghSMFvfnhmyo/Kmvly5XmbI7lksrF8vpsjsrDikrFkpyyo7euvaXcwuj34rh7drwbhw/qq1+7OPwledj7irqvYeI4qj7jrt/4JxXTONnI3z0AAAC+iZkcAAAACGz+tXm9U3tHV396VZ/98jOlZlLyGp4kyTANuZarg6cO+l6iyk/icZzJ96j6HmVCNYprB73GMH1Oou047y/sNSf1bI2j30k9L1G0BwAAQHJQ5AAAAEBoJy+d1MlLJ3X/k/ta/nhZUnOmR+7V0c/cAAAAAACAIgcAAACGljuUU+4QhQ0AAAAAwHhR5AAAAABiIMhGySzFAwAAAABNbDwOAAAAAAAAAAASiZkcAAAAQAwwOwMAAAAAgmMmBwAAAAAAAAAASCSKHAAAAAAAAAAAIJEocgAAAAAAAAAAgESiyAEAAAAAAAAAABKJIgcAAAAAAAAAAEgkihwAAAAAAAAAACCRKHIAAAAAAAAAAIBEosgBAAAAAAAAAAASiSIHAAAAAAAAAABIJIocAAAAAAAAAAAgkShyAAAAAAAAAACARKLIAQAAAAAAAAAAEokiBwAAAAAAAAAASCSKHAAAAAAAAAAAIJEocgAAAAAAAAAAgESiyAEAAAAAAAAAABKJIgcAAAAAAAAAAEgkihwAAAAAAAAAACCRUpMeAAAAALozDGP9ved5oc4L+1n7Of36BgAAAABgkpjJAQAAEEOtAkSrwNBekPB7XqtA0Xp1fjbo+r36BAAAAAAgLpjJAQAAEFOtAkRngSLseUHatQokfq9XvFXU8q1lSVLhcEH5hXzX8/a+53t4z7h7dnDbu2cHX6fbNfq169Wnn76CGLafoPfVq924+xtVn1F/b+3XG3SNoDEd9rke13cR9TM66mdtmD4BAACQLBQ5AAAANqjOIoXfZaeCLFF1ZfGKbl+9rfSmtGRIetrMqTo6f6ZxgwAAIABJREFU8MYBLV5eDDrs0Pa+Fzzp3a/doDZSNEnTYfvp1T5sPPoJE8dBbcO269U26u/Nb6zCxnQYUcd0FL+FOD1rAAAA2JgocgAAAGxQncUKP8ULvwWOpZtLOn/svLI7s9pzdI8ysxkZpqGG05BdtmWtWLpz7Y7OTZ/TmRtnVDhSkDQ4Eeon+dwvCTso+dkrId6r3aDxDurPr7D99JphEDQeQZPCQeM47Di79dl53UHnBu0vrKhiOkjYmIZtN2xMx/WsRdEnAAAAkoU9OQAAAPAMwzDWX63/t1u6uaSLP7mo/EJe+360T/OvzatwuKDcqznt+u4ubd+/XXP75rTj5R3K7s7qwusXdO/X90Y6Zj9/1R0kcTvMZ0FF0U/neX6Sv2GT6kHj2O88v0sx9Tovyu80TP+d157EckhBYxqm3TAxncSzNqnvAgAAAJNBkQMAACCmehUZwp7np137RuXte3a0O3/svLa/tF35hbwKhwt68QcvKv/9vHZ9b5e2vbRNc3vntGV+i2Zzs9qc36z0C2m9f/T9QGObhKT9tbffAsC476uzv7iO0684JsvDxjTp30WnpIwTAAAAo8VyVQAAADHU2k+jVYDotexUv/P67cnRr10/VxavKLsrqx0v79DOV3Yqt5DT3N451e26Kg8qSs2kZBiGGm5Ddbsu13LlVB1ZTyx9cOoDnfjFiZARwfMmyn1Phul/Uv3FsbgCAAAAxBFFDgAAgJjqVXjoPN6vQBH2s17n3L56W3uO7lF2d1ab5zdrbu+ctu7ZKmvFUt2uy6k6sku2MrOZ5iubUTqb1sy2GX16+dORFTkmnZBOej9JEEURIOgm4pMsNMRhDH7wjAIAAGDSKHIAAADAl+KtotKb0srMZpTelJaZNlW367JWLLmWK6/xdHbIlCEzZcpMmzIzplKZVPM1k9L9T+4rdyg31Dj6JVV7reG/973uGxUPm6AdVwI6DonuUcaxn17XHqYIEHaD+1EZZzxHJep9aibxrAEAACCZKHIAAADETLFYfOb/+Xx+ose/+uorffbZZ5r9D7OSIRmmIa/hyak4qjyoqG7X5TU82WVbruWq4TbkNTzJe7rfhykZU802yx8vD13kGEYUCdJx/YV9nP+SfxKJ5mGT3X7jOe5761eYa40njs+ANJ5nlKIGAAAABqHIAQAAECPvvvuu9u/fr+PHj3/jM7/LV43i+PpnntRwGnLKjtYeryk1k5JTdWQYhlzLVe1JTU7FkWu5qjv1ZsGj7jWLHhHplVDt9Zff7W16LXkUt2WM4lrgiCqOYfvtPNbvO28XtMARh7i3FzriaNSxmtSzBgAAgOShyAEAABAzn3/+uU6fPv2N44VCoev5oz6+c+dOHT9+XMUtzZkedtnW2sqaph9OyzAM2SVbxlRzs3Gn4qj2pCa7YsupPlvsMExDhSPd+4iCn6T3MAnZ573A0S7OY+sUJp79EuhJ+H5GbZwxeJ7jDAAAAH8ocgAAAMCX/EJeTtWRtWKp+qiq1HRKDbehzGxGZsqU1/DkWq7siq3aak12+Wmho+rKtZqv3KuTW6qqFz8JWwocg8Vx7HEcU9LFIaZxGAMAAADiw5z0AAAAAJAcB944oNJySdWHVZXvl1VaLjVfxZLK98uqPKyo+qiqtb9dU221tj6rw1q1dPDUwUkPP5S4FjgGLdsTl0Rw2HEOO+4w93/3bO9X5zmj1m82ybAxDfvMxOWZAgAAANpR5AAAAIBvi5cX5VSd9aJGabmk0r1moaN8v6zKbyuqPqxq7as1rT1uFjqsVUtqSCcunhjp2PwsMdTr+KSTusP203l/o9qvYNiCSthxdjuvX59JScYPiqc0vpgOajfumMa9eAcAAID4YLkqAAAABHLmxhldeP2CSkslOVVH6WxaqUxKMiWv7qnu1OVW3fUZHJ7n6e2bb0fW/6Bk7KCCRZh+/PxVfVjD9NO+OXW3tkFj0WuTZz9t+wk7zkHtuhnX99av337HO/sNW5AaVUwHfedBYzruZ23YPgEAAJA8zOQAAABAIIUjBb354ZsqPypr5cuV9SWrysXy+myOysOKSsWSnLKjt669pdzC6Pfi6LeMUL/jSU90jmKT9TB9Deov7Difx++u9dkkYhoXG/l7BwAAQLSYyQEAAIDA5l+b1zu1d3T1p1f12S8/U2omJa/hSZIM05BruTp46qDvJar8JC2HTWwGbT+uRGoU/Yzz3ibRNui+GqMQtnAw7HVH1X5cMY378wIAAIDko8gBAACA0E5eOqmTl07q/if3tfzxsqTmTI/cq6OfuQEAAAAAAEUOAAAADC13KKfcIQobAAAAAIDxosgBAAAARCjIZsksqwMAAAAAw2HjcQAAAAAAAAAAkEjM5AAAAAAixOwMAAAAABgfZnIAAAAAAAAAAIBEosgBAAAAAAAAAAASiSIHAAAAAAAAAABIJIocAAAAAAAAAAAgkShyAAAAAAAAAACARKLIAQAAAAAAAAAAEokiBwAAAAAAAAAASCSKHAAAAAAAAAAAIJEocgAAAAAAAAAAgESiyAEAAAAAAAAAABKJIgcAAAAAAAAAAEgkihwAAAAAAAAAACCRKHIAAAAAAAAAAIBEosgBAAAAAAAAAAASiSIHAAAAAAAAAABIJIocAAAAAAAAAAAgkShyAAAAAAAAAACARKLIAQAAAAAAAAAAEik16QEAAAAAG4lhGOvvPc8LfF778V7XGdSHYRh9+wYAAACAjYKZHAAAAEBEWsWHVoGhW8Fi0Hme5z3z6tZ20OcAAAAA8LxgJgcAAAAQoVbhwfO8vgUHP+d1zsgYNEOj9bnfQkfxVlHLt5YlSYXDBeUX8l3P2/uer8t9w92zg9vePRvsmu3X69W2V59B+uq8RtBxBrl20D66tffTNuw9RdXfKPuM4jvvdq1B7YPGdNjfw7i+i6if0bBjHHWfAABgY6DIAQAAACRIv2Wu/C5RdWXxim5fva30prRkSHrazKk6OvDGAS1eXoxyyH3tfW+4xHeQc1qfhUnuRmVU4+sXx7D3NGisUY8zbJ/DxtTvtcKcF6WoYzqKeI7qWRtFnwAAYOOgyAEAAADEUK+iRbfCht8Cx9LNJZ0/dl7ZnVntObpHmdmMDNNQw2nILtuyVizduXZH56bP6cyNMyocKUganFz0k0Dul0wNUujwY9B4/fTX+nwUCdSw4+s1w8DvfYW9p85rjnqc3frsvK6f84P2GUZUMR0kbEzDths2nmHiMok+AQDAxsCeHAAAAMAGYBjG+qv1/3ZLN5d08ScXlV/Ia9+P9mn+tXkVDheUezWnXd/dpe37t2tu35x2vLxD2d1ZXXj9gu79+t5Ixxxm+agwyfEg/d09O9plboYdX7fz/MQkbFI9SAJ80Hl+l2LqdV7Q4376DNJ/53UnsRxS0JiGaTdMPMPGZRJ9AgCAjYMiBwAAABChXkWGsOf50W2z8s6ZHeePndf2l7Yrv5BX4XBBL/7gReW/n9eu7+3Stpe2aW7vnLbMb9Fsblab85uVfiGt94++P/TYovQ8JzL9FgDG/Vfsnf3FdZx+xfEZCxvTpH8XAAAAfrFcFQAAABCR1tJRrcJFrz0z+p3XeW6367f/348ri1eU3ZXVjpd3aOcrO5VbyGlu75zqdl2VBxWlZlIyDEMNt6G6XZdruXKqjqwnlj449YFO/OJEsEBEjCTsxhd074xR9T+p/uJYXAEAAEgKihwAADwH/CZF+21o3K+9n3Z++gc2gl7PeOfxfr+FsJ/1Ouf21dvac3SPsruz2jy/WXN757R1z1ZZK5bqdl1O1ZFdspWZzTRf2YzS2bRmts3o08ufjqzIEWQj8SiSwHEvlsR9fOMUVRFg3M9YWHEYgx9x2nAdAACghSIHAAAbXPtfirf+crxfoaLbea1/uxUtOq/XrV1nHwDGp3irqPSmtDKzGaU3pWWmTdXtuqwVS67lyms8/c1PGTJTpsy0KTNjKpVJNV8zKd3/5L5yh3JDjSPMptF+Pw8q7onkOIzv7tnmd9Zts+dRJp17XXvYIsC4n7EgfSUxiR+H/UgAAABaKHIAAPAc6FwiZ9jzurUZpFdxpZvV/7gqSdq6Z6uv84E4KBaLz/w/n89P7Pj169d17NgxSdJHlz6SDMkwDXkNT07FUeVBRXW7Lq/hyS7bci1XDbchr+FJ3tOCpCkZU802yx8vD13kCCPK5G/c/1I+zuObRBI+ikKAn5iO+956beTeGke3olJcTOIZjfPvAgAAxAdFDgAAMLQwewR0+vTCp7r5z2/q0f/7SIb59aySna/s1N/7b/6efufU70QyVmAU3n33Xe3fv1/Hjx//xmd+l6+K8vg3jnlSw2nIKTtae7ym1ExKTtWRYRhyLVe1JzU5FUeu5aru1JsFj7rXLHpEpFeSctBsgSiXqYprojSu4+u1MfWoN6zuVwjwWwQIUuCIQ9zbCx1xRIEDAADEGUUOAAAwtF7LVfU71u5f/96/1t9+/rfKbMlo98HdmpqZUsNtyF61VVou6d/843+jP//jP9c/uvGPRnYPwLA+//xznT59+hvHC4VC1/NHeby92HL89HF98fMvZJdtra2safrhtAzDkF2yZUw1Nxt3Ko5qT2qyK7ac6rPFDsM0VDjSve8oDEpe90v8Ji2R3U3cxyfFe2zdBI3psM/YRkeBAwAAxB1FDgAAMDHOmqM/3v/HkiG9+PsvajY3q3Q2LXmSU3VkrVrKfpVV9VFVD/79A/2LPf9C/+Sv/8mkhw0kSn4h3/w9rViqPqoqNZ1Sw20oM5uRmTLlNTy5liu7Yqu2WpNdflroqLpyreYr9+r4l6qKQtwTpXEfXz9xHXtcx5VUFDgAAEASUOQAAOA50JpJMWifDb/ndWsTxs8P/lxT01N68fde1M7v7NRsflZT01OqW/X1hGx6U1pTmSkZhqHV/7iqf/Wf/Cvpp6G6A55bB944oDvX7jQLG1PNjcczsxmZaVPypLpTl1N1ZJebhY7WrA5r1dLBUwcnMuZhZ2fEPVEadHx+Z7xM+n7DjjOK5ZrCxDSqaw2r370PG9OwzwwFDgAAkBTmpAcAAABGq1WAaBUuOpeW8nte63j7+9Z5rWNBlqr61f/wK619tabcoZy+deBbyi/kNf+789r9d3dr27e3acvf2aLZ3Kyy38pq085NemHHC8ruzurJ3zyRbgwVEuC5s3h5UU7VUalYUvl+WaXlkkr3SiotN/9f+W1F1YdVrX21prXHa6qt1mStWlJDOnHxxEjHNop9COKeKB12fJ0xG9VeDr2u63f8YcfZ7bw4JuSDGhRPaXwxHdSOAgcAAEgSZnIAAPAc8LthcdANj/183u2zhtvQn//xn2v7/u3aMr9FW/du1Y5Xdmj7t7dr7as1maaphtOQu9ZcQscu28pszigzm9H01mk5/7cj4/f9zzYBIJ25cUYXXr+g0lJJTtVROptWKpOSTMmre6o7dblVd30Gh+d5evvm25H1PyipGlVis72fQX8dP6h9v+NRFCiCjq99tkO3tqO4pzAFlLDjHNSul2G/8zDCxjRsQWpUMR30nQeNZxRxGVefAABg42AmBwAAGLuVL1dklSy9sO0FTW+ZVibbXELHXXObM0OmDJlpU2baVGo69fVrJqX0C2nJkryVcEtkAc+rwpGC3vzwTZUflbXy5UpzNsdySeVieX02R+VhRaViSU7Z0VvX3lJuYfR7cdw9S/IxiH7J7HH2Nai/sOMcps+46zf+ScUUAABgI2AmBwAAG1ilUnnm/9lsVpL04MEDfetb35Ik3bt3T/Pz82N9X7xVlGEamspMSYbk1lytfbUmr+6pUW/IqTqq23V5Da85E8SQDNNoFj9SpkzTVH25PpqgARvY/Gvzeqf2jq7+9Ko+++VnSs2k5DWeLlVnGnItVwdPHfS9RJWfJOk4k+9R9DfqxG8U1w96jWH6nETbcd5f2GuO694m0W8SvvOo2gIAgI2BmRwAAGxQf/Inf6JPPvlEf/M3f7P+avnLv/zL9fd/+qd/Ovb3lQeV5pJUbrOgYa1Y6/sEVB5UZP2tJbtsy11zVbfrariNZiK28XQPEBkyKixXBYR18tJJ/azxM739f72tP/iXf6A/+Jd/oLdvvq2fNX428j04AAAAACBKzOQAAGCD+s1vfqO//uu/1h/90R9947Mf//jH6+//8A//cOzvv/U731qfsVFbran6sCrDNFQr12ROmarbdTkVR7VSTU7Fkbvmyq25qjt1ea6nhteQdvsOBYAecodyyh0a/ZJUAAAAADAqFDkAAMDYFQ4XVLfrslYtVb+qKvVCSo16Q5nZjKbSU2rUn246XrZVe1KTXbblVB25livXduXVPXkF9uRIMsP4eiZOv43r+53n5xqGYYRqB/gVZCNpltUBAAAAokeRAwAAjF1mc0Yv/u6Levz/Pdb05mmZKVMNp6F0Nq2p9JQ8z2vO5qg6sktPCx0le/3/+juS0pO+C4TVKjJ4nifDMLoWIgad19lmUDGj13m9+gYAAAAAJANFDgAAMBH/8H/9h/r5wZ+r8qAiGVK9Vl8vcsiQGm5zNodTcVR7Ult/1Wt16T+f9OgxrFZhoVXACHreoMJEq3jR79p+FW8VtXxrWVJzFlJ+IT/0NbFxMDsDAAAAmCyKHAAAYCK2v7Rdv/9f/77+3T//d/Lqntw1t1nkyEzJMAw16g3V7brc6tfLVtVKNR39747qzxp/JvHH9+ghqtkZVxav6PbV20pvSkuG1p85p+rowBsHtHh5ceg+AAAAAADDocgBAAAm5of//Q+VfiGtX537leyyrczmjKamp2SYhtSQ6k5drtUscriWqx+++0P93n/1e/qzd/9s0kNHjPRbxqqTn9kdSzeXdP7YeWV3ZrXn6B5lZjMyTEMNpyG7bMtasXTn2h2dmz6nMzfOqHCkEOn9AAAAAAD8o8gBAAAm6u//t39f8787rw/+iw9Uvl9Ww21oKtPcl6NhN2SkDM1smdGp/+2U9hzdM+nhImb87MXReU7nnhztlm4u6eJPLiq/kNeOl3couzur9Ka0vIYnp+xobWVN1UdVZWYzKhVLuvD6Bb354ZsjuDMAAAAAgB8UOQAAwMTt+0/36Z8u/VP95n//jb78P7/U0v+zJEl68Qcvat9/tk/f/vG3m7M7sGH43TOj33ndChzdCh69ZnZ0++z8sfPKL+SVX8hr5ys7tXl+s8y0KafiaO3xmqYfTis1nZI5ZUqSSkslvX/0femfDbxlAAAAAMAIUOQAAACxYJiG9v+D/dr/D/ZPeigYsVbRolW46JxZ0bnZeK/z2v/t/LyXfudfWbyi7K6sdry8Qztf2ancQk5ze+dUt+uqPKgoNZNq7hfjPt0vxnLlVB1ZTyzZV2yJLToAAAAAYOwocgAAAGDsehUkBs3MGHQ87PUk6fbV29pzdI+yu7PaPL9Zc3vntHXPVlkrlup2XU7VkV2ylZnNNF/ZjNLZtGa2zci+bfsaDwAAAAAgWuakBwAAAABMWvFWUelNaWVmM0pvSstMm6rbdVkrllzLldd4OqtkypCZMmWmTZkZU6lMSqlMSkpJxn2WVAMAAACAcaPIAQAAgLEpFovPvPod/6u/+qv1z69fvz7S9x9d+kgymsumeQ1PTsVR5UFFpeWSqo+qssu2XMtVw23Ia3iS93TpK1PNwodhSsthowIAAAAACIvlqgAAADAW7777rvbv36/jx49/47Nuy0i1Hxv1++YBqeE05JSbm4ynZlJyqo4Mw5Bruao9qcmpOHItV3Wn3ix41L1m0QMAAAAAMBEUOQAAADA2n3/+uU6fPv2N44VC4RvHvvvd766/by+MjOT96eP64udfyC7bWltZ0/TDaRmGIbtky5hqbjbuVBzVntRkV2w51Y5ih+fJK1DsAAAAAIBxo8gBAACA515+IS+n6shasVR9VFVqOqWG21BmNiMzZcpreHItV3bFVm21Jrv8tNBRdZt7drielJv0XQAAAADA84ciBwAAACDpwBsHdOfanWZhY6q58XhmNiMzbUqeVHfqcqqO7HKz0NGa1WGtWtKBSY8eAAAAAJ5PFDkAAAAASYuXF3Vu+pxKxZIkybVcZbIZmRlThtFcssq1XDnV5rJVtdVas8DRkLQoidWqAAAAAGDszEkPAAAAAIiLMzfOyCk7Ki2VVFou6cnyE5WXyyoVSyrfL6vysKLqw6rWHq+p+rgqz/P09s23Jz1sAAAAAHhuUeQAAAAAniocKejND99U+VFZK1+uqLTcLHaUi+VmkeO3FVUeVlQqluSUHb117S3lFtiMAwAAAAAmheWqAAAAgDbzr83rndo7uvrTq/rsl58pNZOS12iuRWWYhlzL1cFTB3Xi4okJjxQAAAAAQJEDAAAA6OLkpZM6eemk7n9yX8sfL0tqzvTIvcrMDQAAAACIC4ocAAAAQB+5QznlDlHYAAAAAIA4osgBAAAARMgwjPX3nueFOq/XZ2HaAAAAAMBGxsbjAAAAQERahYZWkaG98OD3PMMw5Hne+quzeNGrgNHvMwAAAADYqJjJAQAAAESoVWjoLFD4PW+chYriraKWbz3db+RwQfmFfNfz9r4X7vp3zw5ue/dssGu2X29Q286+h+kr7DX8XjfItcOOK2w8oupvlH0OG9Ne14r6GRv29zCu7yLqZzTI9xBl26DtAQBAMlHkAAAAAGJm1EWPK4tXdPvqbaU3pSVD0tMunKqjA28c0OLlxcj77GXve8Mlvoc5L0z7IOMNc20pXPK537jCxmOYOIQZZ9g+h42p32uFOS9KUcd0FPEcJi6jajvsbxYAAMQfRQ4AAAAgZjr34Yiq0LF0c0nnj51XdmdWe47uUWY2I8M01HAassu2rBVLd67d0bnpczpz44wKRwqSBic0/SQQ+yVTR5WEbF1zmJkonaJIbg+Kp5/kc/vnfuMYNh6d1xz1OLv12XldP+cH7TOMqGI6SNiYhm03bDyH+e1F0bbdJApSAABgvNiTAwAAAHgOLN1c0sWfXFR+Ia99P9qn+dfmVThcUO7VnHZ9d5e279+uuX1z2vHyDmV3Z3Xh9Qu69+t7Ix1TmGVo/BZUwia0B/UzbKLcT2I46DX8FAzCJtWDJMAHned3KaagsY8ipn7677zuJGYHBI1pmHbDxHOYuIRtO+rfLAAAiD+KHAAAAECEWktN9duPo995g9qFdf7YeW1/abvyC3kVDhf04g9eVP77ee363i5te2mb5vbOacv8Fs3mZrU5v1npF9J6/+j7IxlLWM9zstJvInfcf7Xe2V9cx+lXHJ+xsDFN+ncBAADgF8tVAQAAABFpbSLeKlT0Wnaq33n9NiJvP97Ztt9nVxavKLsrqx0v79DOV3Yqt5DT3N451e26Kg8qSs2kZBiGGm5Ddbsu13LlVB1ZTyx9cOoDnfjFiQiiE964krBB929AdCYd+0kXh3jmwpn0cwMAAOKBIgcAAAAQoV77Z3Qe77fPht9r+P3s9tXb2nN0j7K7s9o8v1lze+e0dc9WWSuW6nZdTtWRXbKVmc00X9mM0tm0ZrbN6NPLn46syOEnsTzJJOa4E9H8Rf3Xoop93J+xOI3Bj7g/oxSPAAB4PlHkAAAAADaw4q2i0pvSysxmlN6Ulpk2VbfrslYsuZYrr/F0VsmUITNlykybMjOmUplU8zWT0v1P7it3KDfUOMJsGu3386j1Guu4EtFxSMzePdu8324bTI8y0T2q2MfpGRtnPEclDs9ou0n/ZgEAwGSxJwcAAAAQge985zsqFovrr5b2Y92O/8Vf/MX6sevXr0f+/qNLH0mGZJiGvIYnp+Ko8qCi0nJJ1UdV2WVbruWq4TbkNTzJe7rclSkZU802yx8vDxGZ8Cad/G1thDyOTabjnIxtFTtar3GIIvZ+YjruZ2zQRu6Tfub7ifMz2jLO3ywAAIgPihwAAADAkH72s5/p2LFj8jxv/dXSfqzX8fZjUb9vHpAaTkNO2dHa4zWV75dVWi6p8tuK1r5aU+1JTU7FkWu5qjv1ZsGj7jWLHhHpTD62JyG7JXYnnVDtl4yOOhE96XvtpVeieNQJ5ChiH6TAEYe4x2EM/cQpVr2M8zcLAADiheWqAAAAgAgUCoVQx9s/P378ePTvTx/XFz//QnbZ1trKmqYfTsswDNklW8ZUc7Nxp+Ko9qQmu2LLqT5b7DBMQ4Uj3e8hCv2WRZL6JyeTkHgdJAn3EOexdRM0phv9GRsWMQAAAHFHkQMAAADYwPILeTlVR9aKpeqjqlLTKTXchjKzGZkpU17Dk2u5siu2aqs12eWnhY6qK9dqvnKvDrcfB7pLcvI4rmOP67iSingCAIAkoMgBAAAAbHAH3jigO9fuNAsbU82NxzOzGZlpU/KkulOXU3Vkl5uFjtasDmvV0sFTBycy5kksM9SaVTIOQe/B74yXSSejw44zitiHiWlU1xpWv3sfNqZhn5m4PFP9jPM3CwAA4os9OQAAAIANbvHyopyqo1KxtL4fR+leSaXl5v8rv62o+rCqta/WtPZ4TbXVmqxVS2pIJy6eGOnY4pqgHOU+IcNep3Nso4phr+v6HX/YcYaJfRIS8oPiKY0vpoPaJSGeneK4tw8AABgPZnIAAAAAz4EzN87owusXVFoqyak6SmfTSmVSkil5dU91py636q7P4PA8T2/ffDuy/gclVaNMQvpJJvfqs/0vw0dRPGi/5qC/3u92rN/YBhUABh0PuidKL2HHGTb2w8Q0rLAxDftMjSqmg77zoPEcxbM2qO2of7MAACD+mMkBAAAAPAcKRwp688M3VX5U1sqXK83ZHMsllYvl9dkclYcVlYolOWVHb117S7mF0e/Fcfds/P7Kul/CeNJj7Te2cfY1qL+w44xz7IfVb/yTiulGsZGfGwAAMBgzOQAAAIDnxPxr83qn9o6u/vSqPvvlZ0rNpOQ1PEmSYRpyLVcHTx30vUSVn+ThOJPvUfY5qsToJMY2TJ+TaDu24w89AAAgAElEQVTO+wt7zXHd2yT6TdrzElV7AACQXBQ5AAAAgOfMyUsndfLSSd3/5L6WP16W1JzpkXt19DM3AAAAACBKFDkAAACA51TuUE65QxQ2AAAAACQXRQ4AAAAACCnIRscspwMAAABEj43HAQAAAAAAAABAIjGTAwAAAABCYnYGAAAAMFnM5AAAAAAAAAAAAIlEkQMAAAAAAAAAACQSRQ4AAAAAAAAAAJBIFDkAAAAAAAAAAEAiUeQAAAAAAAAAAACJRJEDAAAAAAAAAAAkEkUOAAAAAAAAAACQSBQ5AAAAAAAAAABAIlHkAAAAAAAAAAAAiUSRAwAAAAAAAAAAJBJFDgAAAAAAAAAAkEgUOQAAAAAAAAAAQCJR5AAAAAAAAAAAAIlEkQMAAAAAAAAAACQSRQ4AAAAAAAAAAJBIFDkAAAAAAAAAAEAiUeQAAAAAAAAAAACJRJEDAAAAAAAAAAAkUmrSAwAAAAAAIAzDMNbfe5431HmGYXzjs37t/PYNAACA0WImBwAAAAAgcVpFhlaBob3oEPS8Xsc8z1t/tZ/T7zMAAACMFzM5AAAAAACJ1CpcDCo09DuvVbDoPB717IziraKWby1LkgqHC8ov5Luet/e9cNe/e3Zw27tng12z/XqD2nb2HaSvbuMOOtYg1w5y/bBjCxuPqPobZZ/DxrTXtaJ+xob9PYzru4j6GR3mtzfq5xQARoUiBwAAAADgudRtiaqoXVm8ottXbyu9KS0Zkp5251QdHXjjgBYvL460/3Z73xsuiTnMeUHbBxlr0Gu3fxYm+dxvbGHjMWisUY8zbJ/DxtTvtcKcF6WoYzqKeA4Tl1G0jeI3CwBhUeQAAAAAADx3ghQ4Os/1s0TV0s0lnT92XtmdWe05ukeZ2YwM01DDacgu27JWLN25dkfnps/pzI0zKhwpSBqc0PSTROyXTB1VIrJ1zSDJ015/xR/lWAfF00/yOczYwsSjs69xjLNbn53X9XN+0D7DiCqmg4SNadh2w8YzbFzCtB3HbxYAwmBPDgAAAADAc8kwjPVX6//dzulWDGnfk6PT0s0lXfzJReUX8tr3o32af21ehcMF5V7Nadd3d2n7/u2a2zenHS/vUHZ3Vhdev6B7v74X/Q22CbOEjd+CStSFiKiSpH4Sw0Gv4adgEDapHiQBPug8v0sx9Tov6HE/fQbpv/O6k0ich30ug7QbJp7DxGXYmI7qNwsAYVHkAAAAAAAkUr/ixKDz2osU7Xt2dLYbNNuj2znnj53X9pe2K7+QV+FwQS/+4EXlv5/Xru/t0raXtmlu75y2zG/RbG5Wm/OblX4hrfePvu/vpsdk1ElLv0n2SS5VFLexdfYX13H6FcfEeNiYJv278ON5uEcAycVyVQAAAACAxGktGdUqXLQXGtoLD/3O66dXYaTXsZYri1eU3ZXVjpd3aOcrO5VbyGlu75zqdl2VBxWlZlIyDEMNt6G6XZdruXKqjqwnlj449YFO/OJE0FBEigTlxhd074xR9T+p/uJYXAEADIciBwAAAAAgkXoVLDqP+ylsBGnT77PbV29rz9E9yu7OavP8Zs3tndPWPVtlrViq23U5VUd2yVZmNtN8ZTNKZ9Oa2TajTy9/OrIih5/E8qST3+NEMedrURUBkvKMxWEMfvCMAoB/FDkAAAAAAIhA8VZR6U1pZWYzSm9Ky0ybqtt1WSuWXMuV13g6q2TKkJkyZaZNmRlTqUyq+ZpJ6f4n95U7lBtqHGE2jfb7+UYTh/u9e7b5nXXbtHmUie5e1x62CBCnZ2yc8RyVODyjABB37MkBAAAAAEiUYrH4zGuY46urq7p+/fr6sWHef3TpI8mQDNOQ1/DkVBxVHlRUWi6p+qgqu2zLtVw13Ia8hid5T5e+MiVjqtlm+ePl4QMUQhKTv2HF+S/5W8WO1mscWptQD7MZtZ+YjvsZG7SRe5yf+Tg/owAQR8zkAAAAAAAkxrvvvqv9+/fr+PHj3/jM7/JVncfbPx/mffOA1HAacsqO1h6vKTWTklN1ZBiGXMtV7UlNTsWRa7mqO/VmwaPuNYseEemVGB00W+B5SKjG9V57bdo86s2cexUCej0r3QQpcMQh7q37i6s4xQoAkoIiBwAAAAAgUT7//HOdPn36G8cLhULX8wcdby+YDPX+9HF98fMvZJdtra2safrhtAzDkF2yZUw1Nxt3Ko5qT2qyK7ac6rPFDsM0VDjSfaxRGJS87pf43QiJ1yTcQ5zH1k3QmG70Z2xYxAAAwqHIAQAAAABABPILeTlVR9aKpeqjqlLTKTXchjKzGZkpU17Dk2u5siu2aqs12eWnhY6qK9dqvnKvDrcfB7pLcvI4rmOP67iSingCQHgUOQAAAAAAiMiBNw7ozrU7zcLGVHPj8cxsRmbalDyp7tTlVB3Z5WahozWrw1q1dPDUwYmMeRLLDPmdVRJFv0GvNc6xDSPsOKNYrilMTKO61rD63fuwMQ37zMTlmeonKb8LAM8nNh4HAAAAACAii5cX5VQdlYolle+XVVouqXSvpNJy8/+V31ZUfVjV2ldrWnu8ptpqTdaqJTWkExdPjHRscd2HoHNcUY5z2MTrKMfm57p+xx92nN3O2wgJ+UHxlMYX00HtkhDPTuP6XQCAX8zkAAAAAAAgQmdunNGF1y+otFSSU3WUzqaVyqQkU/LqnupOXW7VXZ/B4Xme3r75dmT9D0o4RplM9ZNM7tVn+4yCbtcZdpzt1xz01/tRjW2YeIRJFIcd56B2vQwT07DCxjRs4n1UMR30nQeN5yietUFtR/2bBYCwmMkBAAAAAECECkcKevPDN1V+VNbKlyvN2RzLJZWL5fXZHJWHFZWKJTllR29de0u5hdHvxXH3bPySkP0SxpM2zrH162tQf2HHOUyfcddv/JOK6UbxPNwjgORhJgcAAAAAABGbf21e79Te0dWfXtVnv/xMqZmUvIYnSTJMQ67l6uCpg76XqPKTQBxn8j3KPkeVHJ3E2IbpcxJtx3l/Ya85rnubRL9Je16iag8AUaPIAQAAAADAiJy8dFInL53U/U/ua/njZUnNmR65V0c/cwMAAOB5QJEDAAAAAIARyx3KKXeIwgYAAEDUKHIAAAAAAIDYC7KRNMvpAADw/GDjcQAAAAAAAAAAkEjM5AAAAAAAALHH7AwAANANMzkAAAAAAAAAAEAiUeQAAAAAAAAAAACJRJEDAAAAAAAAAAAkEkUOAAAAAAAAAACQSBQ5AAAAAAAAAABAIlHkAAAAAAAAAAAAiUSRAwAAAAAAAAAAJBJFDgAAAAAAAAAAkEgUOQAAAAAAAAAAQCJR5AAAAAAAAAAAAIlEkQMAAAAAAAAAACQSRQ4AAAAAAAAAAJBIFDkAAAAAAAAAAEAiUeQAAAAAAAAAAACJlJr0AAAAAIZhGMb6e8/zAp83qL2fdn76BwAAAAAA0WMmBwAASKxWoaFVXOhWeBh0nud5PYsTftr1aw8AAAAAAEaLmRwAACDRWgUGz/N6FjmCnBemnWEYfQsda4/WdOt/uaU7/8cdPfyrh5KkXd/dpW//+Ns6/I8Pa2Zuxvd4AAAAAADA1yhyAAAAjNDN//mmfvU//krypMzmjF7Y+oK8hqeH//6h7v36nv7t//Rv9cM/+qF+8F/+YNJDBQAAAAAgcShyAAAADKHfLI6rP72q3/zpb7T77+7W7O5ZZbIZyfj/2bvbGDuu+87zv6r70E3eJtnsJsXuS7pbsiVRNkMv1SIVGzYtjmWvgMkoiMnWRhaxcZaaOJvFOMLu7GQXEyiydjXBGpg32mAcO8mIGISgYnFpIOtkIwzXsZjRcmJZtDKQQjtKaIkOdS+fRHf3fapbj/ui+rZaV32f6j6zvx+goOqqOnVOnVuNFs//nvOXXMtVOVdW6WZJpRsl/eWTf6ns32T1hf/whR63HgCA3utkPq1a94iaiwsAAAwfghwAAABdcOqRU3rrL9/S7GdmNXHXhMamxxQfjct3fJWXyiq+W9TIphHFR+My46Z+dPpH8h1f2t3vlgMA0D2r810ZhlHzywKNrmsUHKl1z+qAB4EOAACGH0EOAAAw1CoDFI3ybDR7XSfKvfXdt/QPL/6DZg7O6LaP36bb9tymzbs2S4ZUXiyrcKMQBjdiZpi43A3kOZ7+7k//TvplSTMtNREAgKHSzXxa9QIXUQIa2fNZZc5nJEnp/WlNz02ved3ssy3fWpJ06YnGZS890do9V9+vUdm16o5SpplyrWi3jijPtVa5XtfXzTo7+bm1845Ffb/aKd+Nz6LT72jUNrZSJ3ArI8gBAACG1upvd1Z+rlg9yNHouur9ZspV17Haf/yf/qPGbx/XlpktmvjIhG7be5sm7pqQ9TNL+WxekuTbvlzLlVN05BQc2QVb1oIl50VH+nJ7/QIAwK2u3rJTnViS6tT8KV04fUGJjQnJkLR8G6foaM8jezT/wnyk+0Yx+2x7A9+tXluvvnr3r5xrd8C13To6/Vz1NGprp9sZtc5Ofm7N9lXUPm1Hp/u0G/3ZrXetXp3AekCQAwAADLVmv60Z9Vud9c6vda54vajrP76umYMz2jCxQaNbRzWyZUSxREyxkZhiozHFN8Tfv42G/02OJVX8x6KMUmuzTQAAWE8afemgmSWpah2/fO6yjj9wXKltKc0cnFFyLCnDNOQ7vux8+IWEi2cu6pmRZ3Ts5WNKH0hLajyg2czgY73B1FYCHc2o9U38Zupr9KydaGvUOtp5rtVlWh2Irr5nt9u5Vp3V923m+lbrjKJTfdpI1D6NWq7d/ozSL/36DIFhYPa7AQAAALeSzKsZmQlT8dG4YomYAi9QeaGsXDan8mJZXtmT7/pSEA6wGKYhM27KjJkyE6aMmCFl+v0UAADcuuoFOE4+fFLTc9O643N3aOf9O5Xen9bUvilt/+h2Tdw1ofE7xjV596RSO1I68dAJvfPKO11taysDllG+zV19bZSB9WbLNqsTdUR5rqiD6q0MgDe6rtmlmFoNQHXqc2s1aNePAfdW+zRKuXb6M2q/9OJ3DxhmBDkAAMDQ+cQnPqFf+IVf0N/8zd+sbJL0ta99beWafu2/8PsvyIyZki+5ZVflpbLyV/PKvZNT4VohXJKq6Mgtu/IcT4EXrAy0GIahmBGTbrTYIQAADJHKclLN5NNq5rpW6641S/P4A8c1ceeEpuemld6f1q5P7NL0fdPa/rHt2nrnVo3Pjmvzzs0amxrTpulNSmxI6LmDz3WsbZ3Qyrr+jQbE+7HcUDsG9bmq6xvUdjZrEAfUo/bpsH8WAN7DclUAAGDoPPTQQ/I87wPHn3jiib7vP/zfPqzvfPc7cixH5aWyijeKMuOm7LwtI2bIK3uy87bsvC235IbBDjuc3eF7vvzAVzAebf1wAAAGXafzaVWfq05Svla5tc6fmj+l1PaUJu+e1LZ7tmlqbkrjs+PybE+FawXFR+MyDEO+68uzvZW8WtaSpW8/+m0d/pPD7XVMmxiEvfX1O+9Cv4NDgxhcATA4CHIAAIChFIvFPnBsdHS07/uzn5xV4AWyl2yVbpaUTCUVBIGSuaTMuCnf9eWWXJVzZZVzZTkF5/3BDt+XsZOcHACAW1e/8mnVK3fh9AXNHJxRakdKm3Zu0vjsuLbMbJG1YMmzPTlFR3bOVnIsGW6ppBKphEa3jur1F17vWpCjmYHlfg9+V7dj2OsYFp0KAgzLOzYIbWjGICVcB9YTghwAAAAdtHnXZm2Z3aLiu8WVhOOe4yk5lpSZeG8ZK6cYzvQoL5Vl5+1wCauiK41LwRgzOQAA6JXs+awSGxNKjiWV2JiQmTDl2Z6sBUuu5Srwl2eVxJbzaCVMmUlT8WQ83EbjuvLaFU3dO9VWO6IkjW72fC/1oi2D8LyXngg/s7WSPXdz0LnWvdsNAgzSO9bL/uyWQchHAqwnBDkAAAA67DO/8xn9+X//50qOJWWYhtyyq+RYUrFEOPvEczy5JVd2wVZ5cTnQkbPllBzpF/rceAC3nFpL9zR7XaPy9c43WzfWh2w2+76fp6enO358ZGREr7/+uh544AFJ0tmzZxvuv/T8S5IhGaahwA/kFBwVrhXk2Z4CPwiXmLRc+a6vwA+kYPndNiUjFpbJvJppO8gRxaAM/vbiW/aD/E3+fnwOnQgENNOnvX62WoncK+1YK6g0KPrxjg7y7wXQSwQ5AAAAOuzjRz+uH/7BD5V9LasgCOSUHCVTScWSMcmQAi9Ymc1h58NAR/Hdoj70yQ/prT1vSYwDAuiQ1fkMKvkN6gUq1rqu8t+1kj9X32/1z/XOYf15+umnddddd+nQoUMfONfq8lKNjlfn6Gi0Hx6QfMeXk3dUullSfDQup+jIMAy5lqvy0vISk5YrzwlzaQVeEAY9OqTWIGWj2QL9HtxczwGOWompu52wul4goNkgQCsBjkHo99WBjkFEgAPoL4IcAAAAXfCrZ39Vf/SJP9L1N67Lue29IEflW6KV2RxO0VHpZyWl59L6le/+ip5++ul+Nx3ALaY6kXO71wFRvfnmm3rsscc+cDydTq95fdTjqwMpTe0/dkhv/f5bsvO2SgsljVwfkWEYsnO2jFiYbNwphMtM2oXlJSZXBTsM01D6wNpt6oRGg9f1Bn67PQi6ngMcqw1y29bSap/28x0bBgQ4gP4jyAEAANAl//yv/7m+82vf0Y++/SMVrhYUHw3X7VYguZYrp+QosTGhfb+6T7/wddapArB+/eziz/SDf/cDvfW9t3Tz4k1J0sSdE7rjn9yh+79yv8ZvH+9zC3Erm56bllN0ZC1YKt4oKj4Sl+/6YT6tuKnAD+Ra7y0zuTqXlmuF29S+3i9V1W8EOOob1LYParuGFQEOYDAQ5AAAAOiih//wYR34jQP6z8/+Z13+/y5r4dKCJGn89nHt/tRuffJ//KRu+69u63MrASCaerM+mp0Rcua3zuiHf/RDKZBGx0e19fat8j1fxatFnf+D83rtude0/zf268HffbDTzQdW7Hlkjy6euRgGNmJh4vHkWFJmwpSCMJ/W6mUmK7M6rEVLex/d25c2R11mqNmZIf1exqjVOjrxXL0QtZ2dWK4pSp926l7tqvfs7fZp1HeGAAcwOAhyAAAAdNnU3JS+8B++0O9mAEBX1EpS3uicJP3x5/9Y2fNZTe2b0tiOMSVTSUmSY4VLA1k3w2/Wf//3vq+rr13VY3/xwaWOgE6Yf2Fez4w8o1w2JymccZlMJWUmTRlGuGSVa4XLTJaXyiovlmUtWpIvHT55uKtt62YegurB3WbqGsQAx1rlW32uTtRTXV+j9kdtZ5QcLcMwON7Mkmy96tNG5QhwAIOFIAcAAAAA3MIqCb8bzapo9rpG5Zs9d+KhE7ryX65o9tCsJu+a1Nj0mOKjcXm2p/JiWcV3iypuKio2GpMRN3Tp5Us6+c9OSgciNQ9o6NjLx3TioRPKXc7JKTpKpBKKJ+OSKQXecj6torsygyMIAj1+7vGO1d9oULWTA5urZwWsVW+jgfJa5RqVb0Y7dXTiueodbzUnSi1R29moXC29+Nzq1VnveK1gTau61aeNPvNW+7MT/dKrzxAYJma/GwAAAAAA6I5KYKESuKg1s6LRdZXjq/dX/7xWEKPeuR+d/pEu//Vl7bx/p277uduU3p/Wrp/fpal9U5q8e1JbZrdo887NSu1IKbU9pY2TGzU2Paa3X3pb+rt2ewVYW/pAWkdfPKr8jbwW3l5QLpNTLpNTPptX/kpehasFFa4XlMvm5OQdfenMlzQ11/1cHJee6M7AZb1B32HWy+eqV1ej+qK2s506B1299verTwEMB2ZyAAAAAMAtrNbsiurjzV7XiXPf+53vaevtW7VlZosmPjKh7T+3XZN3T6r0bkm5TE6BH8izPTklJ8yDULDDXAipspzvOtLdNW8NtGXn/Tv1ZPlJnf7iab3xrTcUH40r8JeDgKYh13K199G9TS9R1cwgaS8H39utuxeDvp2oo5fP1Y+yg/C5RQ0ctHvfbpVvpdwwfObAekOQAwAAAADQM/lsXjcv3tTswVlt2LpBI+MjK7k4zLip2EhM8dH4B7cNcSVSCemnklGItqQW0Kwjzx/RkeeP6MprV5R5NSMpnOkxta/7MzcAAEBrCHIAAAAAAHomcz6zEswwYmFCZ2vBkgytJHf2XV8Klpe8Mg2ZMVNmPNyMmKEgU3uWCNBJU/dOaepeAhsAAAwycnIAAAAAwC3m7NmzOn/+/Pu2itXHvv71r68cf/rpp3uyv/CTBZkxM1ySqhwmGc9fySuXyal4rajyQllOwZFrufIcT4EXhMsFLQc9YkZMuhm5a4bG6pwmUa5rVL7d80CzZp9tfgMAIApmcgAAAADALeall15SEAT66le/+oFz991335r7Tz31VE/2xz88Hs7YKLkqL5VVvFGUGTNl522ZMVOe7cnOhzk4nOJ7wQ7f9eW7vjzfkyaa7oqhtDoBfK3k7Y2uq04mX12uOrl8vZ8BAAAGGUEOAAAAAEDPpO9Ly/d8lZfKKt0sKbExocAPlMwlZcZNBV4gp+TIztnhVrDlFt0w2GF7CvxAQfrWH4BfHayoN5ui2euaFSXAkT2fVeb8ct6K/WlNz0233Q7cOkiYDADoNoIcAAAAAICeGZse0+SdkyrdKKmwpSAzbspzPCXHkoolYuEyVrYnu2DLXlrelmd1OCVHmpSU6vdTDLfqgEh1UKPeudVOzZ/ShdMXlNiYkAxJy5c6RUd7Htmj+RfmO9puAACAtRDkAAAAAAD01Gd/97M6/dhpJTYlZBiG3LK7EuSQJN/x5ViO7Lyt8lJZ1pIlO2/LLbnSF/rc+FtAo+WpGi1ddfncZR1/4LhS21KaOTij5FhShmnId3zZeVvWgqWLZy7qmZFndOzlY0ofSHf/oQAAwLpFkAMAAAAA0FO7f3G3bn/gdl36q0sK/HB5qmQqqVgyJhlS4IWzOZzicqBjsaziu0V95PMf0d/d9XcrMwbQe5fPXdbJh09qem5ak3dPKrUjtbLkmJN3VFooqXijqORYUrlsTiceOqGjLx7td7MBAMAtzOx3AwAAAAAA689jf/6YZj49o8WfLiqfySuXySmXySmfzSt/Na/C1YIK1wsq3iiqcKOgjzz4ET36p4/2u9k9U1kyqlGejWav65TjDxzXxJ0Tmp6bVnp/Wrs+sUvT901r+8e2a+udWzU+O67NOzdrbGpMm6Y3KbEhoecOPteTtgEAgPWJmRwAAAAAgL44+hdH9d3f/q5e/fqryl/NKzYSU3wk/Geqa7nyyp5iG2L61L/6lA49fai/je2hSs6MSuCi1vJRja6r3q+VpLx6qapa507Nn1Jqe0qTd09q2z3bNDU3pfHZcXm2p8K1guKjcRmGId/15dmeXMuVU3TC5cZO2RIpOgAAQBcQ5AAAAAAA9M2D/+ZB7f/yfr3y9Vf09nff1rt//64kaeLuCX34wQ/r/n9xvzbv2tznVvZerYTf1cebva6V87XOXTh9QTMHZ5TakdKmnZs0PjuuLTNbZC1Y7y0vlrOVHEuGWyqpRCqh0a2jsi/YddsDAAAQFUEOAAAAAEBfbZndos9/7fP9bgbqyJ7PKrExoeRYUomNCZkJU57tyVqw5FquAn95VknMkBk3ZSZMmUlT8WRc8WRcikvGld4sqQUAANYXcnIAAAAAADAgstns+7bq47lcTmfPnl053qv9zPmMZEiGaYRJxguOCtcKymVyKt4oys7bci1Xvusr8AMpWF4my1QY+DBMKRO5WwAAAGpiJgcAAAAAAAPg6aef1l133aVDhw594Fx13ox+7CuQfMeXk3dUullSfDQup+jIMAy5lqvyUllOwQnzqTheGPDwgjDoAQAA0CUEOQAAAAAAGBBvvvmmHnvssQ8cT6fTK/urgyC92k/vD+u387ZKCyWNXB+RYRiyc7aMWJhs3Ck4Ki+VZRdsOcWqYEcQKEgT7AAAAJ1HkAMAAAAAANQ1PTctp+jIWrBUvFFUfCQu3/WVHEvKjJsK/ECu5cou2CovlmXnlwMdRTfM2eEG0lS/nwIAANyKCHIAAAAAAICG9jyyRxfPXAwDG7Ew8XhyLCkzYUqB5DmenKIjOx8GOiqzOqxFS9rT79YDAIBbFUEOAAAAAADQ0PwL83pm5BnlsjlJkmu5SqaSMpOmDCNcssq1XDnFcNmq8mI5DHD4kuYlsVoVAADoArPfDQAAAAAAAMPh2MvH5OQd5S7nlMvktJRZUj6TVy6bU/5KXoXrBRWvF1W6WVLxZlFBEOjxc4/3u9kAAOAWRpADAAAAAAA0JX0graMvHlX+Rl4Lby8olwmDHflsPgxyXC2ocL2gXDYnJ+/oS2e+pKk5knEAAIDuYbkqAAAAAADQtJ3379ST5Sd1+oun9ca33lB8NK7AD9eiMkxDruVq76N7dfjk4T63FAAArAcEOQAAAAAAQMuOPH9ER54/oiuvXVHm1YykcKbH1D5mbgAAgN4hyAEAAAAAACKbundKU/cS2AAAAP1BkAMAAAAAAPSMYRgr+0EQRLqu1rlm7t1s/QAAYDiQeBwAAAAAAPREJcBQCS6sDjg0e51hGAqCYGWrDlo0CpysLgsAAIYfMzkAAAAAAEDPVIIL1QGKZq+LGpyoBDhakT2fVeb8cr6R/WlNz02ved3ss5GapEtPNC576YnW7rn6fo3KrlV3vTLNPmerbW62nmbv2+pz1SrX6/q6WWe7fVrrXq2+Y1HeyXbLd+Oz6PQ72m6/tFo/cKshyAEAAAAAAIZK1KBHs+VOzZ/ShdMXlNiYkAxJy5c6RUd7Htmj+RfmW25zVLPPtjfw3eq1rdTXDfWeoXIuyuBzveeKGqRq1NZOtzNqne32abP3inJdJ3W6T7vRn/3oF2A9IMgBAAAAAACGSnUejmYDHY3KXT53WccfOK7UtpRmDs4oOZaUYRryHV923pa1YOnimYt6ZuQZHXv5mNIH0pIaD2g2M4Bcb39VlusAACAASURBVDC104GHWt/Eb1RfMwPFnWhno/5sZvC5leeqLtPqQHT1PbvdzrXqrL5vM9e3WmcUnerTRqL2aad/F7r1rvXqdw8YVuTkAAAAAAAA697lc5d18uGTmp6b1h2fu0M779+p9P60pvZNaftHt2virgmN3zGuybsnldqR0omHTuidV97paptaGbSMMtBZfe0gDJI2MzDc6j2aCRhEHVRvNRhU77pmlyyqdV2rx5ups5X6q+/bj/cp6jvdSrl2+rNf/QLc6ghyAAAAAACAnqksGVUvH0e96xqVi+r4A8c1ceeEpuemld6f1q5P7NL0fdPa/rHt2nrnVo3Pjmvzzs0amxrTpulNSmxI6LmDz3WlLVG1knug0YB4J5a+6qVuPFcnVNc3qO1s1iAO0Eft02H/LKTBbhvQSyxXBQAAAAAAeqKSRLwSqKi1fFS96+olIl99vLpsvXKn5k8ptT2lybsnte2ebZqam9L47Lg821PhWkHx0bgMw5Dv+vJsT67lyik6spYsffvRb+vwnxxuv3PaMCgDnYM4AH6r6PeSRP0ODvFu1Uf/YL0jyAEAAAAAAHqmVv6M6uP18mw0e49mz184fUEzB2eU2pHSpp2bND47ri0zW2QtWPJsT07RkZ2zlRxLhlsqqUQqodGto3r9hde7FuRoZmC534PfvTQowZxB0KkgwLC8Y4PQhmbwjgL9QZADAAAAAACsW9nzWSU2JpQcSyqxMSEzYcqzPVkLllzLVeAvzyqJGTLjpsyEKTNpKp6Mh9toXFdeu6Kpe6faakeUpNHNnu+mfgw+D8JA96UnwmdfK8F0Nwe6a9273c9hkN6xXvZnt/Siv4Yl8AP0Ajk5AAAAAABA1+3evVvZbHZlq1h9bK3jP/7xj1eOnT17tuP7Lz3/kmRIhmko8AM5BUeFawXlMjkVbxRl5225livf9RX4gRQsL4VlSkYsLJN5NdNGz0Q3jIO/UQ3ygG4l2FHZeqGSwLqdRNbN9Gmv37FGidwH+Z0f5HcUuNUxkwMAAAAAAHTVU089pUwms+ZyUa0sPVWdm6MT++EByXd8OXlHpZslxUfjcoqODMOQa7kqL5XlFBy5livP8cKAhxeEQY8OqTUw2mi2wHoYUB3UZ62VmLrbCatrBQJqvStraSXAMQj9Xnm+QTVIfQWsRwQ5AAAAAABA16XT6UjHV58/dOhQ5/cfO6S3fv8t2XlbpYWSRq6PyDAM2TlbRixMNu4UHJWXyrILtpzi+4MdhmkofWDtZ+iERoPX9QZ+uz3w2ouB3WEYPB7ktq2l1T7t5zs2DPrRB/Q78H4EOQAAAAAAwLo1PTctp+jIWrBUvFFUfCQu3/WVHEvKjJsK/ECu5cou2CovlmXnlwMdRVeuFW5T+9rLx4G1DfNA7qC2fVDbNazoT2AwEOQAAAAAAADr2p5H9ujimYthYCMWJh5PjiVlJkwpkDzHk1N0ZOfDQEdlVoe1aGnvo3v70uaoyww1OzOk0aBtt5cOanXwuFPP1W1R29mJ5Zqi9Gmn7tWues/ebp9GfWf69U4N8rJdQL+QeBwAAAAAAKxr8y/Myyk6ymVzyl/JK5fJKfdOTrlM+HPhakHF60WV3i2pdLOk8mJZ1qIl+dLhk4e72rZuDmhW3ztKXd0Y4G138LgTzxWlnurjrQaKmm3nWtcN6oB8Kxr1p9S7Pm1UbhD6c5A/S6DXmMkBAAAAAADWvWMvH9OJh04odzknp+gokUoonoxLphR4gTzHk1t0V2ZwBEGgx8893rH6Gw2qdnJAc/WsgLXq7efg6er2NPr2/lrHojxXM4PrtcpHDQxFaWejcrW006dRRe3TqAGpbvVpo8+81f5s510DUBszOQAAAAAAwLqXPpDW0RePKn8jr4W3F8LZHJmc8tn8ymyOwvWCctmcnLyjL535kqbmup+L49IT3RnwrDfo28ggfIu9lnaeq5N1NaovajvbqXPQ1Wt/v/p00Azy7x7QT8zkAAAAAAAAkLTz/p16svykTn/xtN741huKj8YV+IEkyTANuZarvY/ubXqJqmYGIns5+N6purs5wNqJe7d6j3bq7EfZXj5f1Hv2693qRb39el86UR64VRHkAAAAAAAAWOXI80d05PkjuvLaFWVezUgKZ3pM7ev+zA0AANAaghwAAAAAAABrmLp3SlP3EtgAAGCQEeQAAAAAAAwlwzBW9oMgiHRdrXON7t1s3cB610oiaZbiAQBEQeJxAAAAAMDQqQQZKgGG1UGHZq8zDENBEKxs1YGLWsGLeuUAAADQW8zkAAAAAAAMpUoQolGgodZ1vZyBkT2fVeb8cm6H/WlNz033rG6gn5idAQDoNoIcAAAAAAB0yan5U7pw+oISGxOSIWk5ruIUHe15ZI/mX5jva/sAAACGHUEOAAAAAMC6V1mCqhnNLFF1+dxlHX/guFLbUpo5OKPkWFKGach3fNl5W9aCpYtnLuqZkWd07OVjSh9Id+IxAAAA1h2CHAAAAACAda2VAEdFrSTlUhjgOPnwSU3PTWvy7kmldqSU2JhQ4Ady8o5KCyUVbxSVHEsql83pxEMndPTFox15FgAAgPWGIAcAAAAAYChVghONZlXUuy5KgKNR+eMPHNf03LSm56a17Z5t2rRzk8yEKafgqHSzpJHrI4qPxGXGTElS7nJOzx18TvrXkZsBAACwbhHkAAAAAAAMnUrQohK4qJ5ZUZ1svNZ1q/+7+vzqY9Vl6yUvPzV/SqntKU3ePalt92zT1NyUxmfH5dmeCtcKio/GZRiGfNeXZ3tyLVdO0ZG1ZMk+ZUuk6AAAAGgJQQ4AAAAAwFCqNQOj+niz13Xi3IXTFzRzcEapHSlt2rlJ47Pj2jKzRdaCJc/25BQd2TlbybFkuKWSSqQSGt06KvuCXfO+AAAAWJvZ7wYAAAAAAHAryJ7PKrExoeRYUomNCZkJU57tyVqw5FquAn95VknMkBk3ZSZMmUlT8WRc8WRcikvGlfpLbwEAAOD9CHIAAAAAAIbK7t27lc1mV7aK1cey2az+6q/+auXc2bNnu76fOZ+RDMkwjTDJeMFR4VpBuUxOxRtF2XlbruXKd30FfiAFy0tfmQoDH4YpZdrpGQAAgPWH5aoAAAAAAEPjqaeeUiaTWXPJqHrLVPVsP5B8x5eTD5OMx0fjcoqODMOQa7kqL5XlFBy5livP8cKAhxeEQQ8AAAC0jCAHAAAAAGCopNPppo6v/vnQoUNd389uDmeV2HlbpYWSRq6PyDAM2TlbRixMNu4UHJWXyrILtpxiVbAjCBSkCXYAAAC0giAHAAAAAAAdMD03LafoyFqwVLxRVHwkLt/1lRxLyoybCvxAruXKLtgqL5Zl55cDHUU3zNnhBtJUv58CAABguBDkAAAAAACgQ/Y8skcXz1wMAxuxMPF4ciwpM2FKgeQ5npyiIzsfBjoqszqsRUva0+/WAwAADB+CHAAAAAAAdMj8C/N6ZuQZ5bI5SZJruUqmkjKTpgwjXLLKtVw5xXDZqvJiOQxw+JLmJbFaFQAAQEvMfjcAAAAAAIBbybGXj8nJO8pdzimXyWkps6R8Jq9cNqf8lbwK1wsqXi+qdLOk4s2igiDQ4+ce73ezAQAAhhJBDgAAAAAAOih9IK2jLx5V/kZeC28vKJcJgx35bD4MclwtqHC9oFw2Jyfv6EtnvqSpOZJxAAAARMFyVQAAAAAAdNjO+3fqyfKTOv3F03rjW28oPhpX4IdrURmmIddytffRvTp88nCfWwoAADDcCHIAAAAAANAlR54/oiPPH9GV164o82pGUjjTY2ofMzcAAAA6gSAHAAAAAABdNnXvlKbuJbABAADQaQQ5AAAAAABAzxiGsbIfBEGk62qdW328lXIAAGB4kXgcAAAAAAD0RCXIUAkwVAclmrnOMAwFQbCyrRXYqGzV96xXDgAADCdmcgAAAAAAgJ6pBB8aBRpqXRd1BkaUctnzWWXOL+dS2Z/W9Nz0mtfNPhupSbr0ROOyl55o7Z6r79eobHXdzdRVq72ttrObdaxVPsqz9bq+btbZyc+tm+9Yu78PvfosOv2OtvI5dKpPO/k7C/QbQQ4AAAAAADBUoixl1YpT86d04fQFJTYmJEPS8m2coqM9j+zR/Avzke4bxeyz7Q18t3NdK+Uq59odOG23jlrl6/Vjt/qj0+2MWmcnP7duv2Pt6HSfdqM/2+mXKGV78TsLDAKCHAAAAAAAYKhU5+FYPetjtdXnmjl++dxlHX/guFLbUpo5OKPkWFKGach3fNl5W9aCpYtnLuqZkWd07OVjSh9IS2o8oNnMIGK9wdRWAh2tqNyzlcHTRs/aibZGraPWDINm2xalP6rr6kU716qz+r7NXN9qnVF0qk8bidqnUcu1259R+yVK2X599kAvkZMDAAAAAACsG/UCHCcfPqnpuWnd8bk7tPP+nUrvT2tq35S2f3S7Ju6a0Pgd45q8e1KpHSmdeOiE3nnlna62NcoSNs0GVKIMajYzaNuuTtRRfV0zAYOog+qtDIA3uq7ZZYdqXdfq8WbqbKX+6vv2Y+C81T6NUq6d/mynX6KU7cXvLDAICHIAAAAAAICeqSwn1Sjxd63r2kkYXivAIUnHHziuiTsnND03rfT+tHZ9Ypem75vW9o9t19Y7t2p8dlybd27W2NSYNk1vUmJDQs8dfC5yW7phPQ9aNhsA6PUyStX1DWo7mzWI71jUPh32zwLAe1iuCgAAAAAA9EQliXglUFFv2ala19VLRF4dAFnr3FplT82fUmp7SpN3T2rbPds0NTel8dlxebanwrWC4qNxGYYh3/Xl2Z5cy5VTdGQtWfr2o9/W4T853F7HtIlB2Ftfv/Mn9Ds4NIjBFQCDgyAHAAAAAADomVozKaqP10sa3uw9mj134fQFzRycUWpHSpt2btL47Li2zGyRtWDJsz05RUd2zlZyLBluqaQSqYRGt47q9Rde71qQo5mB5X4Pfle3Y9jrGBadCgIMyzs2CG1oxjC9o8PUVqARghwAAAAAAGDdyp7PKrExoeRYUomNCZkJU57tyVqw5FquAn95VknMkBk3ZSZMmUlT8WQ83EbjuvLaFU3dO9VWO6IkjW72fC/1oi2D8LyXngg/s7WSNndz8LjWvdsNAgzSO9bL/uyWQXhHmzVMbQVqIScHAAAAAADoumw2+76t1ePnzp1bOXf27NmO7WfOZyRDMkxDgR/IKTgqXCsol8mpeKMoO2/LtVz5rq/AD6RgeckrUzJiYZnMq5kO9FDrBmXwtxffsh/kb/JXgh2VrRcqSajbSWTdTJ/2+h1rlMh9UN75tQzyO1ptmNoKNIOZHAAAAAAAoKuefvpp3XXXXTp06NAHzkVZvqrj+4HkO76cvKPSzZLio3E5RUeGYci1XJWXynIKjlzLled4YcDDC8KgR4fUGmxsNFug34OU6znAUSsxdbcTVtcKBNR6V9bSSoBjEPq98nyDapD6qpFhaivQLIIcAAAAAACg695880099thjHzieTqfXvL76+OqfVwdL2t3Pbg5nj9h5W6WFkkauj8gwDNk5W0YsTDbuFByVl8qyC7ac4vuDHYZpKH1g7WfohEaD1/UGfrs9mLmeAxyrDXLb1tJqn/bzHRsGw9QHw9RWoBUEOQAAAAAAwLo1PTctp+jIWrBUvFFUfCQu3/WVHEvKjJsK/ECu5cou2CovlmXnlwMdRVeuFW5T+9rLxzGMCHDUN6htH9R2Dath6s9haivQKoIcAAAAAABgXdvzyB5dPHMxDGzEwsTjybGkzIQpBZLneHKKjux8GOiozOqwFi3tfXRvX9rcz2WGBjHA0eyMl34P8EZtZyeWa4rSp526V7vqPXu7fRr1nRmUd6oZw9RWIAoSjwMAAAAAgHVt/oV5OUVHuWxO+St55TI55d7JKZcJfy5cLah4vajSuyWVbpZUXizLWrQkXzp88nBX2zZoeQgGMcBRq3ytnzul1n2bbX/Udq513a0wIN+oP6Xe9WmjcsPQnxXD1FYgKmZyAAAAAACAde/Yy8d04qETyl3OySk6SqQSiifjkikFXiDP8eQW3ZUZHEEQ6PFzj3es/kaDqp0coGxmMHmtOlefb/TN+qjaqWP1bIe1yjYKADQ63mpOlFqitrNRuVp68bnVq7Pe8XrvWCu61aeNPvNW+7Mb71q9sv347IF+YCYHAAAAAABY99IH0jr64lHlb+S18PZCOJsjk1M+m1+ZzVG4XlAum5OTd/SlM1/S1Fz3c3FceoIByFbUG8zuZV2N6ovaznbqHHT12t+vPgUwHJjJAQAAAAAAIGnn/Tv1ZPlJnf7iab3xrTcUH40r8ANJkmEaci1Xex/d2/QSVc0MkvZy8L3dOnsx6NuJOlq9Rzt19qNsL58v6j379Y71ot5hel8I1GC9IMgBAAAAAACwypHnj+jI80d05bUryryakRTO9Jja1/2ZGwAAoDUEOQAAAAAAANYwde+Upu4lsAEAwCAjyAEAAAAAwIAxDGNlPwiClq9rpXyUckCzWkkkzdI6AIAoSDwOAAAAAMAAqQQaKkGG1YGHZq8LgqBhkGKt+zZTDgAAYJAwkwMAAAAAgAFTCTQEQVAzyNHKddUqMzhaKVNL9nxWmfPLeSv2pzU9N932PXHrYHYGAKDbCHIAAAAAALCOVC9RFdWp+VO6cPqCEhsTkiFp+ZZO0dGeR/Zo/oX5tusAAABohCAHAAAAAADrRCcCHJfPXdbxB44rtS2lmYMzSo4lZZiGfMeXnbdlLVi6eOainhl5RsdePqb0gXSHWg8AAPBBBDkAAAAAAFhHqpeoaiXwcfncZZ18+KSm56Y1efekUjtSSmxMKPADOXlHpYWSijeKSo4llcvmdOKhEzr64tFuPAYAAIAkghwAAAAAAAycZnNmtJpbozqY0erMjuMPHNf03LSm56a17Z5t2rRzk8yEKafgqHSzpJHrI4qPxGXGTElS7nJOzx18TvrXTVcBAADQEoIcAAAAAAAMkErQohK4WB2EWB2UaHRd9X4zwYx65U7Nn1Jqe0qTd09q2z3bNDU3pfHZcXm2p8K1guKjcRmGId/15dmeXMuVU3RkLVmyT9kSKToAAEAXEOQAAAAAAGDA1ApIVB9v9rpm66lX7sLpC5o5OKPUjpQ27dyk8dlxbZnZImvBkmd7coqO7Jyt5Fgy3FJJJVIJjW4dlX3Bbqo9AAAArSLIAQBoyepv99X7R3Ct6+qVX2uZhco11efaTZgJAACA5mXPZ5XYmFByLKnExoTMhCnP9mQtWHItV4G/PKskZsiMmzITpsykqXgyrngyLsUl40pzS2oBAAC0wux3AwAAw6N6yYJaaz/Xuy4IgrrfOFy91TsPAEAnVZb8aSb/Qa3rap1rdO9m68b6sHv3bmWz2ZWt4s0331zZP3v2bM/3M+czkiEZphEmGS84KlwrKJfJqXijKDtvy7Vc+a6vwA+kYPn/AU2FgQ/DlDKt9wcAAEAjzOQAALSkeg3odq+rpdUkmLXks3lJ0tj0WNv3AgDcmlYH5yvBhrX+BtW7rrpMdd6E1eWr71mrHNafp556SplMpuaXPfq9r0DyHV9OPkwyHh+Nyyk6MgxDruWqvFSWU3DkWq48xwsDHl4QBj0AAAC6hCAHAGBoNLtU1o//rx/r5X/7sq6/cV2+54dl44Zu23ObPv2/fFr3fOGerrcVADBc2g3iRw1MENBAtXQ6vebx3bt3r+wfOnSo5/vp/WG77Lyt0kJJI9dHZBiG7JwtIxYmG3cKjspLZdkFW06xKtgRBArSvO8AAKDzCHIAAAbOWt9iXSt/x1oDQ8c/c1zX/vaaRsdGNbl7UomNCfmOL2vR0sJPFvSn/92f6pXfe0W/8pe/0tVnAACg03528Wf6wb/7gd763lu6efGmJGnizgnd8U/u0P1fuV/jt4/3uYW4lU3PTcspOrIWLBVvFBUfict3fSXHkjLjpgI/kGu5sgu2yotl2fnlQEfRDXN2uIE01e+nAAAAtyKCHACAW4JbdvV7d/2eAi/Qhz75IY3tGFMilZACySk5Ki+WVbpZUuF6QZkfZvTsHc/qK29+pd/NBgDcgqIuOVWv3JnfOqMf/tEPpUAaHR/V1tu3yvd8Fa8Wdf4Pzuu1517T/t/Yrwd/98F2mw/UtOeRPbp45mIY2IiFiceTY0mZCVMKJM/x5BQd2fkw0FGZ1WEtWtKefrceAADcqghyAABaUhmAaSYxa9R8HFF84+PfkBkz9aFPf0iTuyc1Nj2m+Ehcbtld+cZhIpWQmTBlmIYWLy3qG/u+If03PWkeAGCd6EaA448//8fKns9qat+UxnaMKZlKSpIcK1wayLoZ/p37/u99X1dfu6rH/uKxtp4BqGX+hXk9M/KMctmcJMm1XCVTSZlJU4YRLlnlWq6cYvhulhfLYYDDlzQvidWqAABAFxDkAAA0bXWi1crPFdUJVutdV73fTMLV6mDJ6mte/j9eVuFaQXd89g5t/7nt2vHxHdryoS0K/ECln5VUuFpQLBGTJPmuL9/x5ZU9Lby9IJ2T9MlI3QEAuIV0IojfjQDHiYdO6Mp/uaLZQ7OavGs5iD8al2d7Ki+WVXy3qOKmomKjMRlxQ5devqST/+ykdKDlZgBNOfbyMZ146IRyl3Nyio4SqYTiybhkSoEXyHM8uUV3ZQZHEAR6/Nzj+uZ3vtnvpgMAgFsUQQ4AQEtqDcI0yqHR6HjUcoEX6JX/8xVN3DmhTTs3aXx2XNvu2aaJOydUerckM26GSyeUHNkFW3Y+3JJjSY1uHpXz146MT/RmtgkAYDB1Moi/VjLyegH+euV+dPpHuvzXlzVzcEa3/dxt2rF3hzbv2iwZkrVgqXCtoPhoXIZpKPCDMIhve3r7pbelzZLubr9vgGrpA2kdffGonjv4nKwlS6NbRxVPxmXElt/D5dkclRkcj597XFNzU9J3+t1yAABwqyLIAQAYagtvL6i0UNJte2/T6JbRleSXruWGg1ExQ7FETLFkTPGR+HvbaFzxjXHphqTFfj8FAKDfuhnEj3rue7/zPW29fau2zGzRxEcmtP3ntmvy7kmV3i0pl8kp8AN5dhjId4rvBfPLqbKc7zoEOdA1O+/fqSfLT+r0F0/rjW+9ofhoXIG/HLgzDbmWq72P7tXhk4f73FIAALAeEOQAADSlWCy+7+eNGze+7/ji4qKmp6clSdlstmf7f/v//q1Mw1QsGZOMMAF56d1S+E1Cz5dTdOTZngI/CAeSjPAf30bMkBk3ZZqm/IzfhR4DACC6fDavmxdvavbgrDZs3aCR8ZGVXBxm3FRsJBYG7Ku3DXElUgnpp5JRYKYiuuvI80d05PkjuvLaFWVezUgKZ3pM7Zvqc8sAAMB6Yva7AQCAwff888/rtdde06VLl1a2ikuXLumnP/2pvvOd99Yg6OX+D176gYzYcqLLUphkPH81r1wmp8LVgqyfWbLzttySK8/25Lt++E3DQFIgGTIU5MmCCQAYLJnzmZVgRuXvnLVgKZfJyVq05FqufNcP/5YZhgzTkBkzwwB+3JQRM6RMv58C68XUvVOa+7U5zf3aHAEOAADQcwQ5AAANvfnmmzpz5ow++tGPrmwVH/3oR3XPPffoy1/+8sqxXu7/0q/90sqMDWvBUvF6UblsTrl3cspfyat4oyjrZ5bKubKcoiO35Motu/IcT77ny5cv3RapWwAAQ+7s2bM6f/78+7aK1cd+8pOf6Omnn14514v9hZ8syIyZ4ZJU5TDJeP5KGMQvXiuqvFCWU3DkWuHftMALVoL4hmEoZsSkm+30DtA9ldw2q3PRtHpd1HOt1A8AAIYDy1UBAIZa+r60PCcc/CndLCmxMaHAD2SP2TLjpnwvTH5p52yVl8qyC3YY7LDCYEfgBlK6308BAOiHl156SUEQ6Ktf/eoHzt13333v+/mpp57q6f6bf/bmyizF8lJZxRtFmTFTdt6WGTPl2Z7sfJiDo/J3zXPCGYu+68vzPWmi2Z4AeqcSWAiCYCXQsFZumnrXVZdp9txaPwMAgOFHkAMAMNRGtoxo5/6dWnh7QcnNYdJxz/bCBOQJUwoUDgQV7PcCHXlbTsGRnbelXZKS/X4KAADeL31fWr7nq7z0/iB+Mhf+rQu8QE7JkZ0L/77ZBVtu0Q2DHZVcVGkGcjGYKkGGSgCj3evWKrOWKAGO7PmsMueX843sT2t6bnrN62afbem2Ky490bjspSdau+fq+zUqu1bdrdbXap2t3m+1Zu8d9bmqy/W6vm7W2W6f1rpXq+9YlHey3fLd+Cw6/Y5G/b3p9O8eMKwIcgAAht4v/vtf1Dfv+6aSV8NohWu5K0EOQ4Y815NruXIKjspLZZUXyyrnyvLKnvRLfW48AABrGJse0+SdkyrdKKmwpRAG8Z0wiB9LxMJlrCpB/KXlbXlWh1NypElJqX4/BdAd1UGPVgIXzZY7NX9KF05fUGJjQjIU5nOT5BQd7Xlkj+ZfmG+53VHNPtvewHer17ZSX6t1tnu/yrkog8/1nivqMzRqa6fbGbXOdvu02XtFua6TOt2n3ejPTvZLP/oYGFQEOQAAQ2/bR7fp57/y83rl668o8AO5JVfJVFJm0pRhGgq8cD1zpxTO3qgEOj75P39S/yn+n1b+0QoAwCD57O9+VqcfO63EpoQMw5BbdleCHJLkO74c672/bdaSJTtvyy250hf63Higi5pdgmqt443KXT53WccfOK7UtpRmDs4oOZaUYRryHV923pa1YOnimYt6ZuQZHXv5mNIHwnVPGw1oNjOAXG8wtdXAQyO1vv3drfpa1ag/mxl8jvJclXOtDh5X37Pb7Vyrzur7NnN9q3VG0ak+bSRqn0Yt125/Ru0XAGsj8TgA4Jbwua99Tp/+rU+HSccv57SUWVI+mw+3K3nlr+VVuF5Q4VpB1oKlB558QJ/93z/b72YDAFDT7l/crdsfuF2LlxbDv2+ZnHLvLP83m1P+al7F60WVDNakHQAAIABJREFU3i3J+pml8kJZxXeL+vDnPizd1e/WA/0VZWmqy+cu6+TDJzU9N607PneHdt6/U+n9aU3tm9L2j27XxF0TGr9jXJN3Tyq1I6UTD53QO6+806UnCEWZUdFKmeprow4sd3JwvJmB4Vbv0UzAoJPP3q12NurvVo83U2cr9Vfftx+BsqjvdCvl2unPTvVLN373gGFGkAMAcMv4zJOf0S9/+5flB77ymbxu/sNNLby9oMWfLmrx0mKYtDVh6ov/9xf1qf/1U/1uLgAADT32549p5tMzWvzpovKZfBjgyOTCIP7VvApXCypcL6h4o6jCjYI+8uBH9OifPtrvZgN1VZaMapRno9nr1ioXJbn48QeOa+LOCU3PTSu9P61dn9il6fumtf1j27X1zq0anx3X5p2bNTY1pk3Tm5TYkNBzB59ruZ5uaiX3QKMB8Wa/YT4og6ydfq5Oqa5vUNvZrEH5vFeL2qd8FsCtg+WqAAC3lI/81x/Rv3znX+rN/+dNvXXmLV3+wWVJ0q6f36UPf+7Duuuf3iXDbO0fygAA9NPRvziq7/72d/Xq119V/mpesZGY4iPhP+Vcy5VX9hTbENOn/tWndOjpQ/1tLNBAJZ9GJXBRa/moetfVy8mxVmCkVgLz1eVOzZ9SantKk3dPats92zQ1N6Xx2XF5tqfCtYLio3EZhiHf9eXZy/neio6sJUvffvTbOvwnh9vvnDYMUv4DdEe/v7nf7+AQA/rv4XcP+CCCHACAW44RM7T74d3a/fDufjcFAICOePDfPKj9X96vV77+it7+7tt69+/flSRN3D2hDz/4Yd3/L+7X5l2b+9xKoDm1ZlnUy58R9R7Nnr9w+oJmDs4otSOlTTs3aXx2XFtmtshasOTZnpyiIztnKzmWDLdUUolUQqNbR/X6C693LcjRzGBmPwa/+zXgzuDuezoVBBjUd2wQ29CMbr+jw9IPQK8R5AAAAACAIbBldos+/7XP97sZwC0nez6rxMaEkmNJJTYmZCZMebYna8GSa7kK/OVZJTFDZtyUmTBlJk3Fk/FwG43rymtXNHXvVFvtiJI0utnz3TCIicl73YbZZ9dOMN3Nge5a92538HuQ3rFe9me3dLO/BuH9BwYNQQ4AAAAAANB1u3fvVjabXfl5enpakt53rNfHE4mEXnr+JcmQDNNQ4AdyCo4K1wrybE+BH8jO23ItV77rK/ADKVheDssMZxAHfqDMq5m2gxxRrKdlqgb5G+z96JNOBAKa6dNeP1utRO6VdqwVVBoU3X5HhzHYA/QKQQ4AAAAAANBVTz31lDKZzJrLRbW69FRXjgeS7/hy8o5KN0uKj8blFB0ZhiHXclVeKsspOGEeHMcLAx5eEAY9OqTWwGij2QLraZmqQRvcrpWYutsJq+sFApoNArQS4BiEfl8d6BhEvQpwDMJnAQwighwAAAAAAKDr0un0QB4/9NghvfX7b8nO2yotlDRyfUSGYcjO2TJiYbJxp+CovFSWXbDlFN8f7DBMQ+kDa9fRCY0Gr+sN/HZrYLSXdQ7D4O4gt20trfZpP96xYdLLPuCzANZGkAMAAAAAAKxb03PTcoqOrAVLxRtFxUfi8l1fybGkzLipwA/kWq7sgq3yYll2fjnQUXTlWuE2ta/3S1WtB8M8aDuobR/Udg0r+hMYDAQ5AAAAAADAurbnkT26eOZiGNiIhYnHk2NJmQlTCiTP8eQUHdn5MNBRmdVhLVra++jevrQ56jJDzc4MqVU2Sp1RtHq/dp6rl6K2sxPLNUXp007dq131nr3dPo36zvSyDwbpswAGkdnvBgAAAAAAAPTT/AvzcoqOctmc8lfyymVyyr2TUy4T/ly4WlDxelGld0sq3SypvFiWtWhJvnT45OGutq2beQiq7z0oOQ/aHbTt1XPVum+z7Y/azrWuG6QB+aga9afUuz5tVG4Y+hNYT5jJAQAAAAAA1r1jLx/TiYdOKHc5J6foKJFKKJ6MS6YUeIE8x5NbdFdmcARBoMfPPd6x+hsNqnZyMHX1rIC16u3nwO3q9jT69v5ax6I8VzOD67XKRwmgRG1no3K1tNOnUUXt06gBqW71aaPPvNX+bOddA1AbMzkAAAAAAMC6lz6Q1tEXjyp/I6+FtxfC2RyZnPLZ/MpsjsL1gnLZnJy8oy+d+ZKm5rqfi+PSE90Z8Kw36DvMevlc9epqVF/UdrZT56Cr1/5+9SmA4cBMDgAAAAAAAEk779+pJ8tP6vQXT+uNb72h+GhcgR9IkgzTkGu52vvo3qaXqGpmkLSXg+/drLtT9+rEfVq9Rzt19qNsL58v6j179Wz9qLdf70sv7wkMG4IcAAAAAAAAqxx5/oiOPH9EV167osyrGUnhTI+pfd2fuQEAAFpDkAMAAAAAAGANU/dOaepeAhsAAAwyghwAAAAAAADoilYSSbPsDgAgChKPAwAAAAAAAACAocRMDgAAAAAAAHQFszMAAN3GTA4AAAAAAAAAADCUCHIAAAAAAAAAAIChRJADAAAAAAAAAAAMJYIcAAAAAAAAAABgKBHkAAAAAAAAAAAAQ4kgBwAAAAAAAAAAGEoEOQAAAAAAAAAAwFAiyAEAAAAAAAAAAIYSQQ4AAAAAAAAAADCU4v1uAAAAANZmGMbKfhAELV/XTPlG1xiGUbduAAAAAAD6iZkcAAAAA6gSfKgEGFYHI5q9LgiChsGRyjW1AhwAAAAAAAwyZnIAAAAMqErgIQiCugGHZq9brdEMjcr5Zu9XXiwrcz4jSUrvT2tk80hT5QAAAAAAaAdBDgAAgHWq3jJXzS5R9b3f/p5e/earspYsJTYkJElO0dHollEd+B8O6ND/dqiTTQYAAAAA4H0IcgAAAKxTawU2mg1w5N7J6Q/2/4F8x9fYzjHt+PgOGTFDnuPJKTgq3Cjo+89+Xz/89z/Ul89/WWNTY918FAAAAADAOkWQAwAAAO9TvURVdeBj8aeL+sMDf6iNkxu1fc92jU2NKbExocAP5BQdWQuWNkxuUPF6UUuXl/TNfd/Ur//w13v9GAAAAACAdYAgBwAAwIBqNi9Gq/kz6qmexbHWzI5v7PuGNu/crPSBtLbds02b0psUS8bklBxZNy0VbxQV3xCXGTclQ1r6xyV9/eNfl77SdvMAAAAAAHgfghwAAAADqBK0qAQuauXMaHRd9X6tJOXN5uD4s1//MxkxQ9v3bNf2j23X1L4pbZnZIt/1VXy3qMLGgoyYocAP5Du+vLInt+Rq4e0F6c8l/dMInQEAAAAAQA1mvxsAAACAtQVBsLJVH2/2uuqtmXL16nrt+GuavGtSm9KbtOVDWzRx54Qm757UpulN2jCxQSNbRjSyeUTJTclwGwu3DVs3SD+M0gsAAAAAANTGTA4AAAA05frfXpcZNzW6ZVTJsaTio3EFfiA7Z8tzPBkyZMZMmXFTsURMsWRMsZHlbTQmmZJxo/0ltQBgPWh2tl2965q5x1rLErZSPwAAQL8xkwMAAGDA7Nq1Szdv3lzZKm7evKnLly+v/Pz666/3dD/zakaGaYS5NgLJKTkqXi8ql8mp9G5JdsGWZ3sKvPdmhxiGEZaJmTINU8E7DJQBQCPVSwzWyrlU77pK8KKyrXWPevdtZrYfAADAIGAmBwAAwAD5zd/8TV27dk2Li4srxyYmJiRJi4uLyufz2rVrlyTp7//+77V3796e7c8UZmQYhjzHk12wZf3MUn5DXk7RkQzJtVyVl8pyio68siff8eV7vgL/vQE2w2EmBwA0o1YOpWavaxScqAQyqu9da2ZHPdnzWWXOZyRJ6f1pTc9Nr3nd7LMt3XbFpScal730RGv3XH2/RmXXqrvZ+topG+Xerdw/atuqy/W6vm7W2W6f1rpXq+9YlHey3fLd+Cw6/Y628jl083cPwOAhyAEAADBAtm7dqq1bt6557o477njfz4cPH+7p/j+e+0cFfiCn4Ki8WFbhekEyJDtvy4gZ8h1fdsFWeaksu2DLKa0Kdri+fN9XMMU3ggGgV2oFPRoFMpoNlpyaP6ULpy8osTEhGZKWL3WKjvY8skfzL8xHb3yLZp9tb+C71Wubqa+dslHbtfpclMHnem2LGqRq1NZOtzNqne32abP3inJdJ3W6T7vRn+32Szd/9wAMJoIcAAAAaEp6f1qu5cpatFS8UQxzcnhhTg4zbsr3fLmWKztvq7xYlp235RScMNhheQrcQMZOZnIAQK+sFdhoZqZGo4DI5XOXdfyB40ptS2nm4IySY0kZ5nKwO2/LWrB08cxFPTPyjI69fEzpA2lJjQc0mxl8rDeY2ukBzFrfxG+mvnbKNqtR3c0MPkdpW+VcqwPR1ffsdjvXqrP6vs1c32qdUXSqTxuJ2qdRy7Xbn1H6pRe/ewAGDzk5AAAA0JRYMvb/s3fnQXJch53nf5l1dAPVABoNNFHdDeOgCB6CYBEQSFmahQHrGO5qzBkbAC2InDEdoM/dnUV4Yzzr2F1awQmuHN6NdSxjPB7LVhD2GguKwICO8RHmiqGQYNMUL4jkiIZWdEAkZKC7cbLRdWXluX8kqtloVnVlZd3d309EBQtZ+fK9fJWlCL1fv/d0++dvV2GyoMKVgvLTeeUmc8pdzCk3mVPhUkHFK0WVrpVUer+k8o2yyvlyOPMjV5a2SYHBTA4A6DbDMOZelX9HdeGlCzr+4HGN7RrT1s9t1cT9ExrfPa7svVmN3jOqkW0jGt46rHV3rlNmQ0bHHjimi69ebNetSIq3hE0jZRae26myjVw3Th2Ntu38kfiD6o0MgNc7L+oMmlrnNXo8Sp2N1L/wut0YcI/7XDZSrpn+bEW/tOu3B6A3EXIAAAAgsp879XMqzZRUuFRQbioMN2YnZ8P3UznlL+U/HHTkwn069MVutx4A+kfUAKLWebXKzd9QfP5+HlEd3XtUI3eMaGzXmMZ3j2vjT2zU2CfGNPrRUa29Y62GNw9r9cRqDWWHtGpslVIrUnp6z9ORr98Jjew9UG9AvNpfmDdTtt16tW0L6+vVdkbViwPqcfu0n76LfmorgNZiuSoAAABEls6k9bPHflZ/+St/qcAP5JZcpTIpJdKJcEkTP5Bne3JKTrhs1c2NyB969iGd+PsTc+u1AwBqqywrVQkqai0ftdh5jW5EHqXcyYMnlRnNaN2d67T+7vXK7spqePOwPNtT4XJBycGkDMOQ7/rybE+u5copOrJmLT136Dnt//r+alV2DAObS1+cmTrtqL9b9fViuAIAnUDIAQAAgIZ87NDHZBiG/uxf/ZmsG5YGhweVSCdkmqaCIJDnePIsT9asJddy9dDXH9Ld+++W/r7bLQeA/lErlFh4fLHwIkqwUe2cWuXOnjqrTXs2KbMho1UTqzS8eVhrNq2RNWOFAXfRkZ2zlR5Kh69MWqlMSoNrB/W9E99rW8gRZWC524PfnUSY84FWhQD98oz1Qhui4BkF0GqEHAAAAGjY9i9u15bPbNHxLxzXlb+/It/zZSQMKZACP5CZMHXbx27TI88/ohUjK7rdXABAk6bOTCm1MqX0UFqplSmZKVOe7cmaCQPtwL85qyRhyEyaMlOmzLSpZDoZvgaTmn5jWtmd2abaEWfT6KifLzW9cL/nj4TfWbXNnts50F3r2s2GAL30jHWyP9ulF55RAEsDIQcAAABiyYxm9Euv/ZJm3pvRxZcv6r3T70mStuzboolPTmh4y3B3GwgAfWhqauqWf4+NjUU+nkwmdfbsWe3du1eSdPr06Za9H/rBkGRIhhkuTegUHBUuF+TZngI/kJ235VqufNdX4AdScHNfEFMyEmGZydcnmw454ujHwd+4evkv+bvxPbQiCIjSp52+t1obuVfaUS1U6hW9/IwC6F+EHAAAAGjK8JZhDW8Z1vZD27vdFADoa0888YS2bdumffv2feizOMtXtfx9IPmOLyfvqHS9pORgUk7RkWEYci033Iep4Mi1XHmOFwYeXhCGHi1Sa2C03myB5TCg2qv3Wmuz53ZvAr1YEBA1BGgk4OiFfp8fdPSiXuorAEsLIQcAAAAAAD3inXfe0cMPP/yh4+Pj41XPX3h8fkDSyvdTq8MZI3beVmmmpIErAzIMQ3bOlpEINxt3Co7Ks2XZBVtO8dawwzANjd9X/R5aod7g9WIDv0th4LUf7qGX21ZNo3261J+xZtEHANqJkAMAAAAAACxqbNeYnKIja8ZS8WpRyYGkfNdXeigtM2kq8AO5liu7YKt8oyw7fzPoKLpyrfCVvbfzS1UtB/08eNyrbe/VdvUr+hNAu5ndbgAAAAAAAOh92x/artxkTsUrReWn88pN5sLXVE756bwKVwoqXi2q9H5J5RvluVkd1g1LOw7t6Eqbzx+p/Vp4zsJyUrwNrJsp26hGr9XJtjUjbju71adxnrF2WGw2SbN9GveZ6eQz1S/PN4DWI+QAAAAAAAB1HTxxUE7RmQs1cpM55S6GQUd+Oq/CpYKKV4oqXSupdD0MOqwbluRL+4/vb2vb2rkPwcJrN1JXM2WjXjvugG072xblulHbH7ed1c7rpQH5uOr1p9S5Pq1Xrpv92annG0BvYLkqAAAAAAAQyeEXD+vYA8eUu5CTU3SUyqSUTCclUwq8QJ7jyS26czM4giDQYy891rL66w1UtnIwdf4mztXqXayuZspGMf+aUf56vxVtizK4Xqt8nAHmuO2sV66WZvo0rrh9GnfAvl19Wu87b7Q/m3nW2v3bA9CbmMkBAAAAAAAiGb9vXI88/4jyV/OaeW9mbsmq/FR+bjZH4UpBuamcnLyjR194VNld7d+Lo13LAS026NvOsu3WybYtVle9+uK2s5k6e129cK0bfdpr+qmtAFqDmRwAAAAAACCyifsn9Hj5cZ360im9/ezbSg4mFfiBJMkwDbmWqx2HdkReoqqZwKAZje5j0c46unHdRq/RrX6IW7aT9xf3mp26t27U2+3fDYEGsLwQcgAAAAAAgIYdeOaADjxzQNNvTGvy9UlJ4UyP7L3tn7kBAABQQcgBAAAAAABiy+7MKruTYAMAAHQHIQcAAAAAoC8ZhjH3PgiChs9brPz8zxotCzSqkY2kWYYHAIBbsfE4AAAAAKDvVEKGSsBQLZSod14QBDUDispn1c4xDOOWz2rVDQAAgPZjJgcAAAAAoC9Vwod6QUPU82qphBrNmDozpckzN/et2D2usV1jTV0PSwuzMwAAiI+QAwAAAACABiwMSxYLQE4ePKmzp84qtTIlGZJunuoUHW1/aLsOnjjY5tYCAAAsbYQcAAAAAADUUG0Wx8Jj1c658NIFHd17VJn1GW3as0npobQM05Dv+LLztqwZS+deOKcnB57U4RcPa/y+8Y7cDwAAwFJDyAEAAAAAQAtdeOmCjj94XGO7xrTuznXKbMgotTKlwA/k5B2VZkoqXi0qPZRWbiqnYw8c0yPPP9LtZgMAAPQlQg4AAAAAQF+qzKCot89G1PNa5ejeoxrbNaaxXWNaf/d6rZpYJTNlyik4Kl0vaeDKgJIDSZkJU5KUu5DT03uelv7njjQPAABgSSHkAAAAAAD0nUpoUQkuai0fVe+8he/rLUM1/5rz/11x8uBJZUYzWnfnOq2/e72yu7Ia3jwsz/ZUuFxQcjApwzDku74825NruXKKjqxZS/ZJW2KLDgAAgIYQcgAAAAAA+lKtDb8XHo96XiOf1/rs7Kmz2rRnkzIbMlo1sUrDm4e1ZtMaWTOWPNuTU3Rk52ylh9LhK5NWKpPS4NpB2WftRdsDAACADzO73QAAAAAAAJaCqTNTSq1MKT2UVmplSmbKlGd7smYsuZarwL85qyRhyEyaMlOmzLSpZDqpZDopJSVjujNLagEAACwVhBwAAAAAgL4yNTV1y2vh8ddee23u2OnTpzv2/tvPfFsyJMM0wk3GC44KlwvKTeZUvFqUnbflWq5811fgB1Jwc5ksU2HwYZjSZJweAQAAWL5YrgoAAAAA0DeeeOIJbdu2Tfv27fvQZ5UlpHzf/9CxTrwPD0i+48vJh5uMJweTcoqODMOQa7kqz5blFBy5livP8cLAwwvC0AMAAAANI+QAAAAAAPSVd955Rw8//PCHjo+Pj9/yX0m3hCFtf//wPr37H9+VnbdVmilp4MqADMOQnbNlJMLNxp2Co/JsWXbBllNcEHYEgYJxwg4AAIBGEHIAAAAAANACY7vG5BQdWTOWileLSg4k5bu+0kNpmUlTgR/ItVzZBVvlG2XZ+ZtBR9EN9+xwAynb7bsAAADoL4QcAAAAAAC0yPaHtuvcC+fCYCMRbjyeHkrLTJlSIHmOJ6foyM6HQUdlVod1w5K2d7v1AAAA/YeQAwAAAACAFjl44qCeHHhSuamcJMm1XKUzaZlpU4YRLlnlWq6cYrhsVflGOQw4fEkHJbFaFQAAQEPMbjcAAAAAAICl5PCLh+XkHeUu5JSbzGl2clb5ybxyUznlp/MqXCmoeKWo0vWSiteLCoJAj730WLebDQAA0JcIOQAAAAAAaKHx+8b1yPOPKH81r5n3ZpSbDMOO/FQ+DDkuFVS4UlBuKicn7+jRFx5VdhebcQAAAMTBclUAAAAAALTYxP0Terz8uE596ZTefvZtJQeTCvxwLSrDNORarnYc2qH9x/d3uaUAAAD9jZADAAAAAIA2OfDMAR145oCm35jW5OuTksKZHtl7mbkBAADQCoQcAAAAAAC0WXZnVtmdBBsAAACtRsgBAAAAAECPMQxj7n0QBE2dZxjGhz6LWw4AAKDXsPE4AAAAAAA9pBJAVAKG+YFEo+fVOhYEwdwrajkAAIBexEwOAAAAAAB6TCW4qBVCRDmvEmYsPF5vdkatcrVMnZnS5Jmb+43sHtfYrrGq521+KtLlPuT8kfplzx9p7Jrzr1evbLW6G6mv2fKNXLeRa8dt18Jyna6vnXU226e1rtXoMxbnmWy2fDu+i1Y/o838zuNeA0B/IOQAAAAAAGCJibvUVCPlTh48qbOnziq1MiUZkm4Wc4qOtj+0XQdPHGy4/rg2P9XcwHej50apb7F6GmlvnGtL8QafF2tX3JCqmX6I2/9x6my2T6NeK855rdTqPm1HfzbbL+387QHoTYQcAAAAAAAsIY0EFfPPjVruwksXdHTvUWXWZ7Rpzyalh9IyTEO+48vO27JmLJ174ZyeHHhSh188rPH7xiXVH9CMMvC42GBqqwcva/0lfqP1NToAHFW9/owy+BznviqfNXoPC6/Z7nZWq3PhdaOc32idcbSqT+uJ26dxyzXbn3H7ZbH6uxEsAWg/9uQAAAAAAGCJMQxj7lX5d7Vzqm1Ivli5Cy9d0PEHj2ts15i2fm6rJu6f0PjucWXvzWr0nlGNbBvR8NZhrbtznTIbMjr2wDFdfPVim+4yFGfpqEbKLDy3kWV9ap3b7EB5lIHhRq8RJTCIO6jeyAB4vfOizqBptO9b0adR6l943W7MKojzTDdarpn+bKZf2v3bA9CbCDkAAAAAAOgxi4UT9c6bv6n4/D07FpZbeCxKuaN7j2rkjhGN7RrT+O5xbfyJjRr7xJhGPzqqtXes1fDmYa2eWK2h7JBWja1SakVKT+95OkYPtE8rQ4p++6vwXr2vhfX1ajuj6sWB9Lh92u/fBYDlgeWqAAAAAADoIZVNvyvBxfygYX44sdh5i6kVjNRz8uBJZUYzWnfnOq2/e72yu7Ia3jwsz/ZUuFxQcjApwzDku74825NruXKKjqxZS88dek77v74/Wge0SacGYePMFkFrdLvvux0OLfdnrtvfP4DuIeQAAAAAAKDH1Aodqs2+aPRaUcOQheedPXVWm/ZsUmZDRqsmVml487DWbFoja8aSZ3tyio7snK30UDp8ZdJKZVIaXDuo7534XttCjigDy90c/Oz0QDR/Uf+BVvV9rz9jvdSGKDodOFb0er8AiI+QAwAAAAAALGrqzJRSK1NKD6WVWpmSmTLl2Z6sGUuu5Srwb84qSRgyk6bMlCkzbSqZToavwaSm35hWdme2qXbE2TQ66uetVqutnRqI7oUB3fNHwvuttsF0Owe629X3vfSMdbI/26Vd/dXt3x6AziPkAAAAAIAlLOqSRLXOq1c+bjlUd9ddd2lqamru32NjY5J0y7Gox2/cuKG7775bknT69Gnt3bs39vuhHwxJhmSYhgI/kFNwVLhckGd7CvxAdt6Wa7nyXV+BH0jBzWfAlIxEWGby9cmmQ444uj34243B/V4cxO3G99CKvo/Sp52+t1obuVfaUS1U6hWdfEaXQhAEIBo2HgcAAACAJWrhXg21NrFe7Lz5m1BXKzd/o+qo5VDdl7/8Ze3du/dDm39L1TcFr3d8vlqfNfQ+kHzHl5N3VLpeUn46r9xkToVLBZWulVSeLcspOHItV57jhYGHF4ShR4ucP1L9JVUfwOz2oH+twWip9QOu3b7XWuZ/R1GOt7LeWsei9n0jAUcv9HsvtGEx3Qw45h8j7ACWHmZyAAAAAMAStnCT6mbPq1YGrTM+Pt6y4/M/27dvX1Pvp1aHM0bsvK3STEkDVwZkGIbsnC0jEW427hQclWfLsgu2nOKtYYdhGhq/r3pbW2GxZZGkxQc1e2mQOq5+uIdebls1jfbpUn/GmkUfAGgnQg4AAAAAkbDsEappxffz/rn39dp/eE3vfutdXT93XZI0cseItv7UVt3/r+/X8JbhlrQV8Y3tGpNTdGTNWCpeLSo5kJTv+koPpWUmTQV+INdyZRdslW+UZedvBh1FV64VvrL3dn6pquWgnwePe7XtvdqufkV/Amg3Qg4AAAAAdc1fzsgwjLlliho5b7ElkxZeL2o5dF+t7y2qF/7tC/ru174rBdLg8KDWblkr3/NVvFTUmT88ozeefkO7f223PvuVz7a66WjQ9oe269wL58JgIxFuPJ4eSstMmVIgeY4np+jIzodBR2VWh3XD0o5DO7rS5rjLDEWdGVJvb4R2a3TwuJn76qS47WxF38fp01Zdq1mL3XvqrW72AAAgAElEQVSzfRr3men0ElUsRQUsT+zJAQAAACCShaFDs+dVK9MqpWslnfvGOZ37xjmVrpdaem20zp9+/k/1xtfeUPberO74r+/Q7Z+9XRP3Tyi7M6vsJ7LKfjyroQ1DeuXfv6Lj/83xbjd32Tt44qCcoqPcVG5uP47cxZxyk+G/C5cKKl4pqnStpNL1kso3yrJuWJIv7T++v61t68Rm3nHqauc+Ic1ep5n7aqaehcfrtT9uO+P0fa+EPIup159S5/q0Xrlu9mcv7tEDoH2YyQEAAACgJ7Ri2aNv/k/f1Ot/9Lrsgq3UYEqS5JZcpYZS2v0ru/XZ315+swEqsyvqzYSJel61MnEce+CYpt+a1uZ9m7Vu2zoNjQ0pOZiUZ3sq3yireK2o4qqiEoMJGUlD5188r+M/fVy6L1Z1aJHDLx7WsQeOKXchJ6foKJVJKZlOSqYUeIE8x5NbdOdmcARBoMdeeqxl9dcbVG3l4OX8vwqvVu9iddUr26z516z31/vVjsW5ryiD67XKx+mDuO2M2/fN9Glccfs07jPVrj6t95032p/NPGvt/u0B6E2EHAAAAAB6QjPLHs3+aFZf3f1VyZeGNw1rxboV4WbIji+7YKt0taTXfv81vfnHb+qXX/9lrZpY1Y5b6Dnzlw2r/Lti4ZJgi5238H2tTcqjlvv+qe/rwssXtGnPJt32sdu0YccGrd64WjIka8ZS4XJBycGkDNNQ4AfyHV+e7em9b78nrZZ0Z9Ndg5jG7xvXI88/oqf3PC1r1tLg2kEl00kZiZvflevLtdy5GRyPvfSYsrvavxdHu/4yu9byN1Hqa6Zsu3Wybd3ow17u+2YttiQTfRrqp7YCaA1CDgAAAAB9bea9GX3tk19TZjSj0e2jGsoOKbUypcAP5jZKLo2UNHhlULMXZ/XVXV/VL732S91udsfUCosWHo96Xtzrz/et3/qW1m5ZqzWb1mjkIyMa/dio1t25TqVrJeUmcwr8QJ7tySk54R4PBTvc5yFTlvNNh5Cjyybun9Dj5cd16kun9Pazbys5mFTg39w7xzTkWq52HNoReYmqqAOzrdaKPRfaWbbd1230Gt3qh7hlO3l/ca/ZqXvrRr3d/t0QaADLCyEHAAAAgEh6ddmjP7j3DzS8aVjj941r/d3rNTQ2pEQ6IbfkqvR+ScUrRaUGU+HmyIY0+4+z+urOr0r/Q6zq0KT8VF7Xz13X5j2btWLtCg0MDyidSUuSzKSpxEBCycHkh18rkkplUtKPJKPAJvS94MAzB3TgmQOafmNak69PSgpnemTvbf/MDQAAgApCDgAAAAB19eqyR3/+i3+uRDKh0e2jGv3oqLL3ZrVm0xp5rqfS1ZJS06m5JY88x5NruXItVzPvzUh/KemfNd83aMzkmcm5MMNIGPJdX9aMJRmaW+rId30pCL9vwzRkJkyZyfBlJAwFk63dqB7Nye7MKruTYAMAAHSH2e0GAAAAAOgPQRDMvRYej3rewlez5d784zc1cueIhsaGtHrjaq39yFqNbBvRquwqDY4MamDNgAZWDyi9Kh2+htJKZ9JaMbxC+m4reqU37du3Tw8++KDOnDkz96p44oknuvp+5oczMhNmGDyVw03G89N55SZzKl4uqjxTllNw5FquPMdT4AXhUkg3Q4+EkZCuN94nQC/b/FT0FwAAuBUzOQAAAAD0pctvX1YyndTgmkGlh9Jz+wLYOVue48nQBzMAEqmEEumEEgM3X4PhLILgytKcEbB3796an335y1/u6vvh24fDGRslV+XZsopXizITpuy8LTNhyrM92flwDw6n+EHY4bu+fNeX53vSSP0+AAAAwPJAyAEAAABgUdeuXbvl3+vWrZMkvfXWW/r4xz/etfdTZ6YkUzIShhRITslR8UpRvuPL933ZeVuu7YYzAW7O/pi//JFhGDIm2duh08Y/MS7f81WeLat0vTS3SXw6l5aZNBV4gZySIztnh6+CLbcYLjPm2Z4CP1AwvjTDKSxfbJIMAEB8LFcFAAAAoKannnpKr7zyinK53Nyr4t133+3qe7toy1C4p4NdsFV6v6T8dF6zk7MqXCqo9H5J9mw4G8Are2H44fnhIHkQyJChwGawvNOGxoa07o51Kl0tqXClMLdUVW4yp/xUXvnpvIpXiipeK8p635I9+8GsDqfkSOskZbp9FwAAAOgVzOQAAAAAUNPMzIxeffVVfeELX/jQZz/zMz/T1fdj944pCAI5BUfWjKXi5aIMw5ibEeA5npyCo/JsORwkLznyLE++fXPZo8CTMc5Mjm74zFc+o1MPn1JqVUqGYcgtu0oPpZVIJSRJvuPLsRzZeVvl2bKsWSucmVNypZ/tcuMBAADQUwg5AAAAAPSl8d3jcqww4ChdKyk5mJTv+UoP3Vz2yA/kWu7cQLmds2UXbTlWGHbIEcsedcld//wubdm7Ref/5rwCP1yeKp1JK5FOSIYUeIE825NTvBl03CireK2oj3z+I/rBth9IfG0AAAC4ieWqAAAAAPQlM2Xqjn96h/JTeRUuF1SYLig/mb9l2aPC5YJK10oqvV+aCzoqszt0pyQmcnTNw3/1sDb9V5t040c35r63ue/uUl6FSwUVrhRUvFpU4WpBH/nsR3ToPx/qdrMBAADQY5jJAQAAAKBvffHPvqivZL6iwuWCJMm1XKWGUuGyR4bku75cyw2DjVxZ5RvlMOwo2hLj5V33yF8/om/+L9/U67//uvKX8koMJJQcCP9vqmu58sqeEisS+ie/8U+074l93W0sAAAAehIhBwAAAIC+lRxM6qGvP6Q//6U/V+AFcks3Q450QoZhhMdsV27p5rJVubJcy9UXn/uinn3rWZY96gGf/d8+q92/vFuv/v6reu+b7+naP1yTJI3cOaLbP3u77v/v79fqjau73EoAAAD0KkIOAAAAAH3tnofukZEwdPLQSZVnyxpYM6DEQEKGaUi+5LmeXMtVebYsp+Toi6e+qLv++V3SW91uOSrWbF6jz//O57vdDAAAAPQhQg4AAAAAfe/u/Xfr31z6Nzr+heOafnNaQRCEMzkUSL5kmIayu7L6l3/9LzWweqDbzQUAAADQIoQcAAAAAJaEFWtX6LHvPKbZC7O6+MpFnf+b85KkzXs3a+L+CZY8AgAAAJYgQg4AAAAAS8rqjau1euNq3XPgnm43BQAAAECbmd1uAAAAAAAAAAAAQByEHAAAAAAAAAAAoC8RcgAAAAAAAAAAgL5EyAEAAAAAAAAAAPoSIQcAAAAAAAAAAOhLhBwAAAAAAAAAAKAvEXIAAAAAAAAAAIC+RMgBAAAAAAAAAAD6UrLbDQAAAAAAIA7DMObeB0HQ8Hnzj7fqMwAAAHQWIQcAAAAAoO9UgoYgCGQYhgzDqBo21DtvsYAi7mcAAADoHEIOAAAAAEBfqgQNlQCj2fPaaerMlCbPTEqSxnePa2zXWNXzNj8V7/rnj9Qve/5IY9ecf71aZWvVGaWuZspG1Wwd1crHubdO19fOOlv5vUV5xmrV2+j5C8Up347votXPaDt/e534zQJAHIQcAAAAAIBla7Elr+J+Nt/Jgyd19tRZpVamJEPSzVOdoqPtD23XwRMHm2h9YzY/1dzAdyPnVD5rNByJUjaqZuuoVX6xfowbUtVra6vbGbfOVn5vUfsqbp82o9V92o7+bNezVqvOTvxmASAuQg4AAAAAwLJULbiYP+sjzmcVF166oKN7jyqzPqNNezYpPZSWYRryHV923pY1Y+ncC+f05MCTOvziYY3fNy6p/oBmlEHExQYoGwk6oqjX3nr1tbutcdtXa4ZBo/fV6ED0wmu2u53V6lx43SjnN1pnHK3q03ri9mnccq36DTXSL83U2cn/fQGAqMxuNwAAAAAAgKXkwksXdPzB4xrbNaatn9uqifsnNL57XNl7sxq9Z1Qj20Y0vHVY6+5cp8yGjI49cEwXX73Y1jY1MvAYNVBpNLxY+Hkjg6hxNNO+Wuc1c1+1LNbf7Wpnve84znfTjmesck43Bs4b7dM45dr1G6pXLu5n7f7NAkBchBwAAAAAgL5UWTKq3j4bUc9rlaN7j2rkjhGN7RrT+O5xbfyJjRr7xJhGPzqqtXes1fDmYa2eWK2h7JBWja1SakVKT+95uiNti2o5D1pGDQA6vYzSwvp6tZ1R9eIzFrdP+/27AIB+x3JVAAAAAIC+U9lEvBJczF8uauHSUoudt/CazX528uBJZUYzWnfnOq2/e72yu7Ia3jwsz/ZUuFxQcjApwzDku74825NruXKKjqxZS88dek77v74/Vn+0CoOwS1+390/odjjUi+EKAKA5hBwAAAAAgL5Ua8PvhcejnteKz86eOqtNezYpsyGjVROrNLx5WGs2rZE1Y8mzPTlFR3bOVnooHb4yaaUyKQ2uHdT3TnyvbSFHIxuJt2IQuJmB7E4MwhPmfKBVIUCnn7G4eqENUfTShutRy/V6nwJYugg5AAAAAABogakzU0qtTCk9lFZqZUpmypRne7JmLLmWq8C/OaskYchMmjJTpsy0qWQ6Gb4Gk5p+Y1rZndmm2hFn0+ionzcqyvWqtbdTg6W9MCh7/kjYB9U2bW7nQHetazc7YN3pZ6yRuvox3OqF/UgW6uZvFgCqIeQAAAAAAPSVu+66S1NTU3P/Hhsbk6RbjjV7fGZmRvfcc48k6fTp09q7d2/d90M/GJIMyTANBX4gp+CocLkgz/YU+IHsvC3XcuW7vgI/kIKbS1+ZkpEIy0y+Ptl0yBFHKwd/W/FX3dUG/Full//qvBuD8K0IAqL0aafvrdZG7pV2tPMZa1Y3ntFm6+zl/gSw9BFyAAAAAAD6xpe//GVNTk5WXTKq0WWp4ix3Vfd9IPmOLyfvqHS9pORgUk7RkWEYci1X5dmynIIj13LlOV4YeHhBGHq0SK2BxnqzBVq5TFUj16p2bq22NqtXA45aG1O3e8PqxYKAqP3fSMDRC/0+P+joRf0QcHTyNwsAURByAAAAAAD6yvj4eNuPz/9s3759kd5PrQ5nhth5W6WZkgauDMgwDNk5W0Yi3GzcKTgqz5ZlF2w5xVvDDsM0NH5f9Ta1Qr3B68UGfjs9kN3oQHsUvTTQXksvt62aRvu02WdsqeuHgKOWdvxmASAqQg4AAAAAAFpgbNeYnKIja8ZS8WpRyYGkfNdXeigtM2kq8AO5liu7YKt8oyw7fzPoKLpyrfCVvbfzS1W1Qq8PUPd6+xbTq23v1Xb1q34OOACg2wg5AAAAAABoke0Pbde5F86FwUYi3Hg8PZSWmTKlQPIcT07RkZ0Pg47KrA7rhqUdh3Z0pc3Nzs7o9YHSOEvxRJnx0u37jdvOVizX1IrljeJeq1mL3XuzfRr3mSHgAIDmmN1uAAAAAAAAS8XBEwflFB3lpnLKT+eVm8wpdzGn3GT478KlgopXiipdK6l0vaTyjbKsG5bkS/uP729r29qxD0HcgdJ6bWlVW1uxmfJi/26VWteN2v647ax2Xi8OyDeqXn9KnevTqM96rwccnfrNAkAczOQAAAAAAKCFDr94WMceOKbchZycoqNUJqVkOimZUuAF8hxPbtGdm8ERBIEee+mxltVfb7Cx1Xtc1Kuz3mD5YpppazPtmz/boVrZRu+p1obiUcouJm4765WrpdnvPI64fRp30L1dfVrvO2+0P1vRL62qs145AGg3ZnIAAAAAANBC4/eN65HnH1H+al4z782Eszkmc8pP5edmcxSuFJSbysnJO3r0hUeV3dX+vTjOH+mdAch67eiFti42mN3JuqL0VSPHW1Fnr1us/d3q037XD79ZAMsXMzkAAAAAAGixifsn9Hj5cZ360im9/ezbSg4mFfiBJMkwDbmWqx2HdkReoirK4GEnB99bUV+7B0Rbcf1Gr9FMnd0o28n7i3vNTt1bN+rth++8VWUBoJ0IOQAAAAAAaJMDzxzQgWcOaPqNaU2+PikpnOmRvbf9MzcAAACWA0IOAAAAAADaLLszq+xOgg0AAIBWI+QAAAAAAAA9r5GNpFlWBwCA5YONxwEAAAAAAAAAQF9iJgcAAAAAAOh5zM4AAADVMJMDAAAAAAAAAAD0JUIOAAAAAAAAAADQlwg5AAAAAAAAAABAXyLkAAAAAAAAAAAAfYmQAwAAAAAAAAAA9CVCDgAAAAAAAAAA0JcIOQAAAAAAAAAAQF8i5AAAAAAAAAAAAH2JkAMAAAAAAAAAAPQlQg4AAAAAAAAAANCXCDkAAAAAAAAAAEBfIuQAAAAAAAAAAAB9iZADAAAAAAAAAAD0JUIOAAAAAAAAAADQlwg5AAAAAAAAAABAXyLkAAAAAAAAAAAAfYmQAwAAAAAAAAAA9CVCDgAAAAAAAAAA0JeS3W4AAAAAAKB9DMOYex8EQcPnzT9e7RqLXT9q3QAAAEBczOQAAAAAgCWqEjJUAoaFgUXU84IgmHstLDf/s/nlFvsMAAAAaBVmcgAAAADAElYJJuoFDVHPq1am0c9qmTozpckzk5Kk8d3jGts1VvW8zU81fGlJ0vkj9cueP9LYNedfr1bZWnVGqauZslE1W0e18nHurdP1tbPOVn5vUZ6xWvU2ev5Cccq347to9TPazt9eJ36zAIBbEXIAAAAAABbV7mWnTh48qbOnziq1MiUZkm5W4RQdbX9ouw6eONjyOmvZ/FRzA9+NnFP5rNFwJErZqJqto1b5xfoxbkhVr62tbmfcOlv5vUXtq7h92oxW92k7+rNdz1qtOjvxmwUAfBghBwAAAACgplpLVC1U6/hin1146YKO7j2qzPqMNu3ZpPRQWoZpyHd82Xlb1oylcy+c05MDT+rwi4c1ft+4pPoDmlEGERcboGwk6IiiXnsXq6+Zsu1uX60ZBlHbVvms0YHohddsdzur1bnwulHOb7TOOFrVp/XE7dO45Zrtzzj9ErfObn33ALCcsScHAAAAAKApcQOO4w8e19iuMW393FZN3D+h8d3jyt6b1eg9oxrZNqLhrcNad+c6ZTZkdOyBY7r46sW23kcjA49RA5UoA6/tKBtVK+pYeF6U+4o7qN7IoHK986IuO9RoANWq763R0K4bA+eN9mmccs3+huL0SzvaQ7ABAO1DyAEAAAAAS1hlqal6+2xEPa9auUYDDkk6uveoRu4Y0diuMY3vHtfGn9iosU+MafSjo1p7x1oNbx7W6onVGsoOadXYKqVWpPT0nqcbalu7LedBy6gBQKeXUVpYX6+2M6pefMbi9mm/fxcAgN7FclUAAAAAsERVNhGvBBfzA4f5AUS98xZec+Fn1fbsWOyzkwdPKjOa0bo712n93euV3ZXV8OZhebanwuWCkoNJGYYh3/Xl2Z5cy5VTdGTNWnru0HPa//X9TfZMcxiEXfq6vX9Ct8OhXgxXAACohZADAAAAAJawWjMpFh6Pel4rPjt76qw27dmkzIaMVk2s0vDmYa3ZtEbWjCXP9uQUHdk5W+mhdPjKpJXKpDS4dlDfO/G9toUcjWwk3opB4GYGsjsxCE6Y84FWhQCdfsbi6oU2RNFLG663qxwAoD5CDgAAAABAx0ydmVJqZUrpobRSK1MyU6Y825M1Y8m1XAX+zVklCUNm0pSZMmWmTSXTyfA1mNT0G9PK7sw21Y44m0ZH/bxRzVyvE4PQvTDQff5I+J1V27S5nYPHta7dbAjQ6Weskbr6cTC+F/YjaXc5AEBthBwAAAAAsARNTU3d8u+xsbGWHj99+rT27t3b8PvJM5OSIRmmocAP5BQcFS4X5NmeAj+QnbflWq5811fgB1Jwc8krUzISYZnJ1yebDjniaOXgbzOD5J34K/te/kv+bgzCtyIIiNKnnb63Whu5V9pRLVTqFd14RuPW2cu/JwBYCgg5AAAAAGCJeeKJJ7Rt2zbt27fvQ581uixVlOONvlcg+Y4vJ++odL2k5GBSTtGRYRhyLVfl2bKcgiPXcuU5Xhh4eEEYerRIrcHGerMFWrlMFQFHY2ptTN3uDasXCwKihgCNBBy90O/zg45eRMABAJiPkAMAAAAAlqB33nlHDz/88IeOj4+PVz2/0ePzA5RG3o/vDq9n522VZkoauDIgwzBk52wZiXCzcafgqDxbll2w5RRvDTsM09D4fdXb1Ar1Bq8XG/ht90D2cg445uvltlXTaJ82+4wtdQQcAICFCDkAAAAAAB0ztmtMTtGRNWOpeLWo5EBSvusrPZSWmTQV+IFcy5VdsFW+UZadvxl0FF25VvjK3tv5papagYCjfXq17b3arn5FwAEAqIaQAwAAAADQUdsf2q5zL5wLg41EuPF4eigtM2VKgeQ5npyiIzsfBh2VWR3WDUs7Du3oSpubnZ2x1AKOqDNeuj3AG7edrViuKU6ftupazVrs3pvt07jPDAEHAKAWs9sNAAAAAAAsLwdPHJRTdJSbyik/nVduMqfcxZxyk+G/C5cKKl4pqnStpNL1kso3yrJuWJIv7T++v61ta8c+BEst4KhVvta/W6XWdaO2P247q53XiwPyjarXn1Ln+rReOQIOAMBimMkBAAAAAOi4wy8e1rEHjil3ISen6CiVSSmZTkqmFHiBPMeTW3TnZnAEQaDHXnqsZfXXG1Rt1QDl/Hrq/XV8K8tG1Uwd82c7VCtbLwCod7zRPVFqidvOeuVq6cT3tlidix1fWGfcQKpdfdrq30Er+qWROrvx3QMAmMkBAAAAAOiC8fvG9cjzjyh/Na+Z92bC2RyTOeWn8nOzOQpXCspN5eTkHT36wqPK7mr/XhznjzAA2YjFBrM7WVe9+uK2s5k6e91i7e9WnwIAEAczOQAAAAAAXTFx/4QeLz+uU186pbeffVvJwaQCP5AkGaYh13K149COyEtURRkk7eTge7P1dWLQtxV1NHqNbvVJ3LKdvL+41+zUvXWj3n74zpstBwBoDiEHAAAAAKCrDjxzQAeeOaDpN6Y1+fqkpHCmR/be9s/cAAAAQH8j5AAAAAAA9ITszqyyOwk2AAAAEB0hBwAAAAAAS0QjG0mztA4AAFgK2HgcAAAAAAAAAAD0JWZyAAAAAACwRDA7AwAALDfM5AAAAAAAAAAAAH2JkAMAAAAAAAAAAPQlQg4AAAAAAAAAANCXCDkAAAAAAAAAAEBfIuQAAAAAAAAAAAB9iZADAAAAAAAAAAD0JUIOAAAAAAAAAADQlwg5AAAAAAAAAABAXyLkAAAAAAAAAAAAfYmQAwAAAAAAAAAA9CVCDgAAAAAAAAAA0JcIOQAAAAAAAAAAQF8i5AAAAAAAAAAAAH0p2e0GAAAAAEC3GYYx9z4IgobPW6z8/M/qfb5Y3QAAAAA+jJkcAAAAAJa1SshQCRgWhhJRzguCYNGAovJ5tYBj/me16gYAAABQHTM5AAAAACx7lfChXtAQ9bxG6412svTe6ff07jff1YWXL0iSNn56o7b+1FZt2btFIh8BAADAMkTIAQAAAABt1uySVP/4d/+o//Sl/yRrxpJhGkqvTCvwA114+YJe/t2XNTgyqC+e+KLGPzneymYDAAAAPY+QAwAAAADaqNYSVQvVOv53//vf6W/+3d9o+PZhbdixQemhtAzDkFt2ZedslWZKKlwq6OhnjupzX/mcPnnkk227FwAAAKDXEHIAAAAAQJfVCjhO/7vTevl3X9bGT2/UyB0jGsoOKbUiJd/1Vc6VVbpe0sDVAaVWpJRckdS3/tdvySk6XbiD9mnlpvCNfNZI3QAAAOgeQg4AAAAAy14lZKi3z0bU8+LUvdDMuzN68bdf1I99+se04cc3aPSjo1o9sVpGwpCds1W8VlQ6k1YilZAkBX4g3/X17Se+Lf13kla1rIldM3+zd8MwFp0Fs9h59TaFr1X3wkCEoAMAAKD3EHIAAAAAWNbmD4xX/l0xf2C73nkL31c+jzKToNqMgT/7+T/Tmh9bo+Gtw1p35zplP57V2tvXys7byl/Ky0gaCrxAnu3JtVy5JVdOwZE9Y6v4XFF6tMmO6RF9sSn8TVNnpjR5ZlKSNL57XGO7xqqet/mpeG06f6R+2fNHGrvm/OvVK1ut7qj1LSzbaDsbuXajdcS9r7j31Kr62llns31a61qNPmNxnslmy7fju2j1M9rI99DO3x4A9ApCDgAAAADLXq0B7YXHo57X7Gd23taFly9o055NWrl+pVaOrlTmtowG1w5KhpTKp5RemVZqZeqD14qUkiuTSq1OST+SDLd1s0363WLLTrViSaqTB0/q7KmzSq1MSYakm5dxio62P7RdB08cjHXdODY/1dzAd6Pn1qsvbqAT1WLXr3wWp32L3Vfce6rX1la3M26dzfZp1GvFOa+VWt2n7ejPZvqlG30KAN1CyAEAAAAAPWbqzJQSA4kwuBhIypChcr6swqWCPNuT7/jhgLwhGaYhI2HITJlKpBJKpBIyEob8i363b6MnLLbxe7Obwl946YKO7j2qzPqMNu3ZFG4KbxryHV923pY1Y+ncC+f05MCTOvziYY3fNy6p/oBmlAHkxQZTGwk6oqj1l/iN1Ff5vB0Dr/X6M8rgc5z7intPC6/Z7nZWq3PhdaOc32idcbSqT+uJ26dxyzXbn838ftr52wOAXmF2uwEAAAAA0A1btmzRZz7zGb3zzjtzr4pz587pT/7kT+b+3en3l/7LJZmmKRmS53gq58KAIzeZU+FqQeUbZTlFJww8PF+BH9wSepimKeMSMzlaYbGA4/iDxzW2a0xbP7dVE/dPaHz3uLL3ZjV6z6hGto3MLTWW2ZDRsQeO6eKrF9va1jhL2DRSZuG5UcOYdi6PE2VguNFrRAkM4g6qNzIAXu+8qEsx1Tqv0eNR6myk/oXX7cYySnGe6UbLNdOfzfRLt/oUALqBkAMAAADAsvToo49q586dGhsbm3tVbNiwQXv37p37d6ffr1y/Ur7ny7Vc2TlbpWsl5afzYcgxXVDxWlHlG2XZeVtuyf1gdocbKPDCwCNYuTQ2ya62b0kz5zVad60lrI7uPaqRO4nOSj4AACAASURBVEY0tmtM47vHtfEnNmrsE2Ma/eio1t6xVsObh7V6YrWGskNaNbZKqRUpPb3n6Za1rRUa2Xug3oB4v/2VeK/e18L6erWdUfXiIHvcPu337wIAljKWqwIAAABaKOr6/rXOi7p3QKNlUd2qVauqHh8aGtLQ0NDcv7ds2dLR99ed6wqCIAw4rpeUHkqH/15lh0tRub7ckqvybFnlXFlOwQk3Hy+78hxPvu/LmOj/mRyt3hQ+zmfVflcnD55UZjSjdXeu0/q71yu7K6vhzcPybE+FywUlB5MyjPB7qmwM7xQdWbOWnjv0nPZ/fX/8TmkBBmGXvjgzddpRf7fq68VwBQDQPoQcAAAAQIvMH2CtDLjWWt+/1nmV/9YLNBaes7Cuxf4CHb1veOuw0kNpWdctFa8WlUgn5Lu+yrmyEsmEAj+QW3blFJy5oMMu2HJKjpySIw1IGu72XbRGr20KL0lnT53Vpj2blNmQ0aqJVRrePKw1m9bImrHk2Z6coiM7Zys9lA5fmbRSmZQG1w7qeye+17aQI8rAcrcHvzuJMOcDrQoB+uUZ64U2RMEzCgCtQcgBAAAAtNDCvyxv9rxaFoYYjQQapaslnfmjMzr3jXO68v9dkSSN3jOqj/zTj2j3r+zW4NrBhtuD1jKTpu77tfv0nd/9jlJDKRmmIc/2lMqklEgnpCDcq8MtubLztso3yirPhstXOXlH+pQUGIRc7TB1ZkqplSmlh9JKrUzJTJnybE/WjCXXchX4N8PLhCEzacpMmTLTppLpZPgaTGr6jWlld2abakecTaOjfr7U9ML9nj8SfmfVNphu50B3rWs3GwL00jPWyf5sl154RgGgnxFyAAAAAMvI3/3O3+lvf/tvJUnpobRWrlkp3/d15e+v6OKrF/Xib7+ovV/eq0/9j5/qckux74l9euv/fkuzF2alQHItdy7kqCyF5JbduVkD5Rtlla6XtGp8la7/5HWpzzOOqampW/5d2TOllcenp6e1c+dOSdLp06fn9kZZ7P3QD4bmNngP/EBOwVHhckGe7Snwg3CfFMuV74Ybwiu4OevKlIxEWGby9cmmQ444+nHwN65e/kv+bnwPrQgCovRpp++t1kbulXZUC5V6RS8/owDQbwg5AAAAgD5TbymqWp+f/LmT+uELP1T2x7PKbMgoPZSWDIV7O+TCAfLStZK+9Vvf0vSZaf3s//Oz7bwNRPBrb/+afu/O39PMezPKlDIfhBw3B9g9O5zN4RQcFa8XNTg8qF/9L7+qr/wfX+l205vyxBNPaNu2bdq3b9+HPmt0Wao4y13VfR9IvuPLyTsqXS8pOZiUU3RkGIZcK9wrpbJPiud4YeDhBWHo0SK1BkbrzRZYDgOqvXqvtTambveG1YsFAVFDgEYCjl7o9/lBRy/qpb4CgKWAkAMAAABYQmoFHM8eeFY/+vaPtPknN2tk24hWja1ScjApz/ZUni2reK2ogVUDSg4mZSZMff8/f1/eIU+6pws3gTnpTFq//qNf1x998o90/R+uyzCNMOhIJhQEgXzbl120pUC6bftt+sWXf1Hq//3GJUnvvPOOHn744Q8dHx8fr3p+nOPzP5sfqCz2fmp1ODPEztsqzZQ0cGVAhmHIzn2wKXxlrxS7YMsp3hp2GKah8fuqt6kV6g1eLzbwuxQGXvvhHnq5bdU02qdL/RlrFn0AAK1HyAEAAAC0UCVkqLfPRtTz4tS90A+/8UP98Bs/1KY9m3Tbjtt028du0+qJ1ZIplW+UVbxSnAs3giBQ4AbyHE/v/MU70qCkLS1rImIwEoZ++fVf1lt//JZe+j9f0rV3rslMmZLC2QTr7lynT//Gp/Xxn/94l1u6PIztGpNTdGTNhJvCJweS8l1f6aG0zKQZbgpvubIL4RJidv5m0FF05VrhK3tv55eqWg76efC4V9veq+3qV/QnALQHIQcAAADQIpXQohJczA8c5gcQ9c5b+L7WdearnDu/fOW8b/zGN7R2y1oNbx7WyEdGdNvHbtO6betUer+k/FT+g02srXB/B6fghAO0M2U5/68j/Upz/YLW+PgvfFwf/4UwyLj+D9clSSPbRrrZpGVr+0Pbde6Fc2GwkQg3Hk8PpcPw6ebvySk6t24KX7Bl3bC049COrrQ57jJDUWeGdHvQttF29Mt9xW1nK5ZritOnrbpWsxa792b7NO4z0yvPFAAsRWa3GwAAAAAsJUEQzL0WHo963sLXYtepV65wuaCrP7iqlRtWanDtoAbXDmpgzYDMlKnEQEKJwYSSK5JKDibD/857nxpKSVcko7RE1j9aQka2jRBwdNHBEwflFB3lpnLKT+eVm8wpdzGn3GT478KlgopXiipdK6l0vaTyjbKsG5bkS/uP729r29q5D8HCa/fKngfNDh536r5qXTdq++O2s9p5S2FAvl5/Sp3r03rl+qE/AaCfMZMDAAAAWMKmzkzJTJpKDiaVSCUUeIHKM2XljJx815dnhfsEKAhngRimITNpykyYMlPhf72LXrdvA+g5h188rGMPHFPuQk5O0VEqk1IynZRMKfDCJd/cojs3gyMIAj320mMtq7/eoGorB1PnzwqoVm+Uv+Cvd7wVAUW9v96vdizOfTVzT3EClLjtrFeulmb6NK64fRo3kGpXn9b7zhvtz3Y8a6367QFAL2EmBwAAANCkV155Ra+//rrefPPNuVfFm2++qbNnz+p3fud35o518v2J/3hCZsKUfMktuyrPlpW/FP7leeFyQdYNK9wvoBxujBx4H8wCMQxDpmHKuMZMDmCh8fvG9cjzjyh/Na+Z92bC2RyTOeWn8nOzOQpXCspN5eTkHT36wqPK7mr/Xhznj7Rn0HKxQd9+1sn7WqyuevXFbWczdfa6xdrfrT4FAHQHMzkAAACAJj3//PNKJpP6zd/8zQ99tmNHuP7+7bffPnfsyJEjHXv/4L96UH/xzb+QYzkqz5ZVvFqUmTRl520ZpiHP9mTnbdl5W27JDcMOO5zd4Xu+/MBXMFx9iSxguZu4f0KPlx/XqS+d0tvPvq3kYFKBfzMkNA25lqsdh3ZEXqIqyiBpJwffW1F3uwd+W3H9Rq/RTJ3dKNvJ+4t7zU7dWzfq7bfnBQD6ESEHAAAA0AKO4yiRSHzoeOXY/M8GBwc79n7zpzYr8ALZs7ZK10tKrUwp8AOlV4UbJvueL7fkqpwLN0d2Cs6tYYfvy5hgJgewmAPPHNCBZw5o+o1pTb4+KSmc6ZG9t/0zNwAAAJY7Qg4AAABgCVu9cbXWbF6j4rWiBtYMKJFKyHd9pXNpmUlTCsJlrJxCONOjPFuWnbfDJayKrjQsBUPM5ACiyO7MKruTYAMAAKCTCDkAAACAJe4nf+sn9Ve/+ldKD6VlGIbcsqt0Jq1EOpxd4jme3FK4QXL5xs2gI2fLKTnSP+ty4wEsK41sJM1yPAAAQCLkAAAAAJa8H3/kx/XdP/yupt6YUhAEcizng5DDkAIvCGdzFB3Z+TDoKF0raeOnNurd7e9KTOQAAAAA0KMIOQAAAIBl4BdO/4K+9qmv6cr3rsi57YOQwzANBX4wN5vDKTgqzZQ0/olx/fw3f15PPPFEt5sOYBlhdgYAAGgUIQcAAACwTPzid35Rf/mrf6mzJ86qMF1QckVSicGEFEie5ckpOUquSGrn4Z36wu99odvNBQAAAIC6CDkAAACAZeSn/+Cndf9/e7++8399Rz/62x9p5vyMJGl4y7Du2nOXPv3rn9box0a73EoAAAAAiIaQAwAAAFhmbvvx2/Qvnv4X3W4GAAAAADTN7HYDAAAAAAAAAAAA4iDkAAAAAAAAAAAAfYmQAwAAAAAAAAAA9CVCDgAAAAAAAAAA0JfYeBwAAAAAljDDMObeB0HQ8Hn1yke9PgAAANAOzOQAAAAAgCWqEkBUwof5gUTU84IgqBleGIYx93kQBDWvDwAAALQLIQcAAAAALGGVgKLeLIuo51UrAwAAAHQLy1UBAAAAANru/XPv67X/8Jre/da7un7uuiRp5I4Rbf2prbr/X9+v4S3DXW4hAAAA+hEhBwAAAACgaZWlq6p54d++oO9+7btSIA0OD2rtlrXyPV/FS0Wd+cMzeuPpN7T713brs1/5bIdbDQAAgH5HyAEAAAAAaMpiAceffv5PNXVmStl7sxraMKR0Ji1JcixH5dmyrOuWileLeuXfv6JLb1zSw3/9cCebDgAAgD5HyAEAAAAAS1glgKi3KXjU82qVq+bYA8c0/da0Nu/brHXb1mlobEjJwaQ821P5RlnFa0UVVxWVGEzISBo6/+J5Hf/p49J9DTUBAAAAyxghBwAAAAAsUZXQohJczA8j5ocT9c5b+L7yeeXf88+pfPb9U9/XhZcvaNOeTbrtY7dpw44NWr1xtWRI1oylwuWCkoNJGaahwA/kO74829N7335PWi3pzhZ3BgAAAJYkQg4AAAAAWMJqzbJYeDzqeVE/+9ZvfUtrt6zVmk1rNPKREY1+bFTr7lyn0rWScpM5BX4gz/bklBw5RUd2wZadt1XOlOV80yHkAAAAQCRmtxsAAAAAAFha8lN5XT93XStvW6kVa1doYHhgbi8OM2kqMZBQcjD54deKpFKZlHRNMgqNLZsFAACA5YmZHAAAAACAlpo8MzkXZhgJQ77ry5qxJEPyXV+u5cp3fSkIl7oyTENmwpSZDF9GwlAwWXuWCAAAAFDBTA4AAAAAWGL27dunBx98UGfOnJl7SdLp06fnzvn/2bv7GDnu+87zn6rp7hmxh+TwyezuocnIerR5PFNjSrEdC2QSe/WXd7EkdZCl4ARQWeRhc9Hu3e3ibg+CoEA54O6PXQhJzkk2EO8PQrLFpYBb5MGANrCV1WkvXhHKQgJxq1vGpkP2jEiKmmE/VVdVV90fzR6NWv1QVV3dXTXzfgENz1TXr36/+nY1lXy/8/v9XnjhhbH9vPq3qzJnzPaSVM32JuPVlaoq5Yrq1+tqrjbl1By5lquW05Lf8uV7/nrRY8aYkW7FEYnp6+x1EmTj937n9Xtv4/FB1w+7mTwAAECaMJMDAAAAADaZ48ePDz3+/PPPj+3nhS8stGdsNFw1bzdVv1mXOWPKrtoyZ0y17JbsansPDqf+SbHDcz15rqeW15J2R7nzZNm4UXunENFrH5NB53W36f590L4oG68NAACwWVHkAAAAAADEqvSVkryWp+btphq3Gspuy8r3fOUqOZkZU37Ll9NwZFfs9qtmy6277WKH3ZLv+fJLm2O5qk4RolPACHvesCLGIJ2CSNBCx/LFZZUvliVJpWMlFZeKPc879FK08Vx5dnjbK8+Gu+bG6w1r2913mL56jTvsWMNcO8z1o44tajzi6m+cfY4a037XivsZG/X7MKnPIu5ndJTv3rifUwDpRJEDAAAAABCr+eK89ty7R42bDdV21mRmTLWclnLzOc1kZ9rLWNkt2TVb9u07rzuzOpyGI+2RlJ/2XSTHoKJHv/f6zRrp5fzp87p04ZKy27KSIelOM6fu6PDjh3X6tdPRBx/SoZdGS2KOcl7Ytp33Rkmcjnr9fu0HxTFqPIaNNe5xRu0zzs9sEs9YVHHHdBzxHNd3L2q7OL6zAJKJIgcAAAAAIHa/9L/+ki48eUHZ7VkZhiG36a4XOSTJczw5liO7aqt5uynrtiW7asttuNI/nPLgE6Zf8aJXwaMzcyNIgePq21d19vhZ5ffmdfDRg8rN52SYhjzHk121Za1auvzGZb04+6LOvHVGpYdLkoYnNIMkEAclU8MUOsLoXDNK4nqc4x0WzyDJ543vBx1X2Hj06msS4+zVZ/d1g5wfts8o4orpMFFjGrXdqPGMGpewbSf1nQWQPGw8DgAAAACI3QN//wH93PGf09qVNVWWK6qUK6pcu/O/yxVVP6yqfqOuxkcNWR9baq42Vf+ori988wvSfdMeffp1b0jevWTV1bev6pVvv6LiUlF3f/NuLT6yqNKxkgpHC9r3xX3afd9uLdy9oD3371F+f17nHjunaz++NtYxR1nCJmhBZRxFiDiSpEESw2GvEaRgEDWpHiYBPuy8oEsxhY1/HDEN0n/3daeRNA8b0yjtRonnKHEZtW2Y4wDSjyIHAAAAAGAsnvyzJ3XwGwe19rM1VcvVdoGjXFF1uarqh1XVPqypdqOm+s26ajdruueX79ET/9cT0x52rPoVGYKeF2XjcN/3P/XqHNvo7PGz2n3vbhWXiiodK+nAVw+o+JWi9n1pn3bdu0sLhxa0Y3GH5gvz2l7cruxdWb386MuhxzJOWzlhGbQAMOlllLr7S+o4g0riMxY1pmn/LABgEJarAgAAAACMzVN/8ZT+8n/5S73zf7yj6odVzczOKDPb/n9FXctVq9nSzF0z+oV/9gs68cKJ6Q42Zp2lozqFikHLTvU7b9BG5N0FkDB7cOT35bXn/j3a++BeFZYKWji0oJbdUu16TZm5jAzDkOd6atktuZYrp+7Ium3p9Sde18nvnQwZiXiRhN38pr13wrSLQ0ksrgBAkjGTAwAAAAAwVr/8u7+sX/+bX9cjv/2I9ty7R67lyrVc7b5/t37+n/y8fvO939x0BY6O7hkVG48HOW/YNQa169fXpQuXtOveXcrvz2v74nYtHFrQzoM7tW3vNs0tzGl2x6xy87lPXvmcsvms5nbN6b3X3gsbgsDCrLk/iSTwsL9sH/dYKOZ84tBLn36Ncp2g50yz0DDqfU5K0sY47e8sgOlhJgcAAAAAYOx2Htqpb/1v35r2MLa85YvLym7LKjefU3ZbVmbWVMtuyVq15FqufO/OrJIZQ2bGlJk1ZeZMZXKZ9msuo5V3V1R4qDDSOKJsGh30/ThdeXZwwnmSxZZp2hiHfhuPj8O4ktVJe8Y2SlrhIIgkPKMdSfjOApg8ZnIAAAAAABCz5eXlT73CHr948eL6e2+++WZsP5cvliVDMkxDvufLqTmqXa+pUq6ofrMuu2rLtVx5riff8yX/zrJYpmTMtNuU3ynHEKHw0pj8jSrJf3Ee16yKMDqbUI+yGXWQmE76GRu2kXuSn/kkP6MAth5mcgAAAAAAEKMXXnhB9913n06cOPGZ9/otKzVoOavYf/Ylz/HkVB01bjWUmcvIqTsyDEOu5ap5uymn5rT3THFa7YJHy28XPWLSLzE6bLbApBOqg/rtN9Zx9jlN/ZYCGveG1f0KAWHiH6bAkYS4d+4vqZIUq27T+M4CmD6KHAAAAAAAxOyDDz7Qk08++ZnjpVKp5/ndxzf+vrFYMurPyzvas0fsqq3GakOzN2ZlGIbsii1jpr3ZuFNz1LzdlF2z5dQ/XewwTEOlh3vfQxyGJa8HJX7jTrwOu17YRHscfSZBksfWS9iYTvIZS6Mkx2Aa31kAyUCRAwAAAACALaK4VJRTd2StWqrfrCszm5HnesrN52RmTPmeL9dyZddsNdeasqt3Ch11d33D+MLR0fbjQG9JTh4Pk9SxJ3VcaUU8ASQVRQ4AAAAAALaQw48f1uU3LrcLGzPtjcdz8zmZWVPypZbTklN3ZFfbhY7OrA5rzdKRJ45MZcxpWWYoqrD3EHTGy7RjEnWccSzXFCWmcV1rVIPufdSYRn1mkvJMAUAvbDwOAAAAAMAWcvq103LqjirLFVVXqqqUK6pcq6hSbv9e+7Cm+o26Gh811LjVUHOtKWvNkjzp5Csnxzq2JO5D0G9McY111ORx9zjGFcNhcRg2/qjj7HXeZkjIB3muJhXTYe3SEM+Nxv2dBZA8zOQAAAAAAGCLOfPWGZ177JwqVyty6o6y+awyuYxkSn7LV8tpya276zM4fN/XM28/E1v/w5KNcSZTgyY8B80mGPaX9XGMLWwfw8Y3rAAw7HjYPVH6iTrOoPHvNkpMo4oa06hJ93HFdNhnHjae43jWBrWdxHcWQDIxkwMAAAAAgC2m9HBJT/3gKVVvVrX609X2bI5yRdXl6vpsjtqNmirLFTlVR0+/8bQKS+Pfi+PKs8lKQA4aTxLGOmhsk+xrWH9Rx5nk2I9q0PinFdPNIOnfWQDjwUwOAAAAAAC2oMVHFvVc8zld+M4Fvf/995WZy8j3fEmSYRpyLVdHnjgSeImqIMnDSSbf4+xzXInRaYxtlD6n0XaS9xf1mpO6t2n0m7bnJa72ANKFIgcAAAAAAFvYqVdP6dSrp7Ty7orK75QltWd6FI6Of+YGAADAqChyAAAAAAAAFR4qqPAQhQ0AAJAuFDkAAAAAAIllGMb6z77vhz5vUPuN7w27PhBEmI2kWU4HAIB4sPE4AAAAACCROkWITvGhuygR5Dzf9wcWLzrvU+AAAABIJ2ZyAAAAAAASq1N88H2/b5EjzHmjWr64rPLFO/tWHCupuFQcW19IH2ZnAAAweRQ5AAAAAABbVtDlsM6fPq9LFy4puy0rGZLunOrUHR1+/LBOv3Z6zCMFAABALxQ5AAAAAABbUq89OrqPXX37qs4eP6v83rwOPnpQufmcDNOQ53iyq7asVUuX37isF2df1Jm3zqj0cGmStwAAALDlUeQAAAAAAKCHq29f1SvffkXFpaL23L9H+f15Zbdl5Xu+nKqjxmpD9Zt15eZzqixXdO6xc3rqB09Ne9gAAABbCkUOAAAAAEBidWZXDNtnI+h5YZw9flbFpaKKS0XtfXCvti9ul5k15dQcNW41NHtjVpnZjMwZU5JUuVrRy4++LP2L2IYAAACAIShyAAAAAAASqVO06BQuNi4ltXFpqWHndf/ceb+7ILKx3fnT55Xfl9ee+/do74N7VVgqaOHQglp2S7XrNWXmMjIMQ57rqWW35FqunLoj67Yl+7wtsUUHAADARFDkAAAAAAAkVr/NwLuPBz0v6HuXLlzSwUcPKr8/r+2L27VwaEE7D+6UtWqpZbfk1B3ZFVu5+Vz7lc8pm89qbtec7Et2gDsDAABAHMxpDwAAAAAAgCRZvris7LascvM5ZbdlZWZNteyWrFVLruXK9+7MHJkxZGZMmVlTZs5UJpdRJpeRMpKxEt+yWQAAAOiPIgcAAAAAIHEeeOABLS8vr786rl+/rjfffHP993H8/KNXfyQZkmEa7U3Ga45q12uqlCuq36zLrtpyLVee68n3fMm/s/SVqXbhwzCl8ugxAAAAwHAsVwUAAAAASJTnn39e5XI50BJU4/i5fUDyHE9Otb3JeGYuI6fuyDAMuZar5u2mnJoj13LVclrtgkfLbxc9AAAAMDEUOQAAAAAAiVMqlXoe/9znPqfPfe5z67+fOHEi/p+fPKGffPcnsqu2GqsNzd6YlWEYsiu2jJn2ZuNOzVHzdlN2zZZT7yp2+L78EsUOAACASaDIAQAAAADABsWlopy6I2vVUv1mXZnZjDzXU24+JzNjyvd8uZYru2arudaUXb1T6Ki77T07XF8qTPsuAAAAtgaKHAAAAAAAdDn8+GFdfuNyu7Ax0954PDefk5k1JV9qOS05dUd2tV3o6MzqsNYs6fC0Rw8AALB1UOQAAAAAAKDL6ddO68XZF1VZrkiSXMtVLp+TmTNlGO0lq1zLlVNvL1vVXGu2CxyepNOSWK0KAABgIsxpDwAAAAAAgCQ689YZOVVHlasVVcoV3S7fVrVcVWW5oupKVbUbNdVv1NW41VD9Vl2+7+uZt5+Z9rABAAC2FIocAAAAAAD0UHq4pKd+8JSqN6ta/emqKuV2saO6XG0XOT6sqXajpspyRU7V0dNvPK3CEptxAAAATBLLVQEAAAAA0MfiI4t6rvmcLnzngt7//vvKzGXke+21qAzTkGu5OvLEEZ185eSURwoAALA1UeQAAAAAAGCIU6+e0qlXT2nl3RWV3ylLas/0KBxl5gYAAMA0UeQAAAAAACCgwkMFFR6isAEAAJAUFDkAAAAAABgTwzDWf/Z9P/J5/d4Pcn3DMAb2DQAAkGZsPA4AAAAAwBh0ChCdAsPGgkSY8zpFis6r3/Fe1+/XJwAAwGbBTA4AAAAAAMakU5ToV4QYdt6gWRjDZmd02gYtdCxfXFb54p39Ro6VVFwq9jzv0EuBLvcZV54d3vbKs8Ov0+saQdr1u0bYtkGuuVHQ60e9r+52k+5vXH2OGs9B1xt2jbAxHfW5ntRnEfczOs5nLe7PH8DmRpEDAAAAEzfq8i1Rl3UJ2z8AJEGUf7PCLFF1/vR5XbpwSdltWcmQdKeZU3d0+PHDOv3a6bBDjuzQS+GT3sPahblOVMPGJkVLPkeNxyCjxDHKOKP0OWo8w/Qf5bw4xR3Tfu1Giemkn7W4P38Amx9FDgAAAEzUxmVZDMPom4gbdN6gpV+6rzfsdwBIuiD/hm08HvTfuatvX9XZ42eV35vXwUcPKjefk2Ea8hxPdtWWtWrp8huX9eLsizrz1hmVHi5JGp6YDJJ8HJSEHZbc7ZcQD1PoGIdhcQmSfN74fth4hE1Eh43jqOPs1Wf3dYedG7a/qOKK6TBRYxq13agxndSz1q9dmLYAthb25AAAAMDEdRcrRj2vV5teohY41n62prWfrYVuBwCT0Ovftk5xuFMM7i4KX337ql759isqLhV19zfv1uIjiyodK6lwtKB9X9yn3fft1sLdC9pz/x7l9+d17rFzuvbja2O9jyB/tR4mcRv2OqMIm6gNcl6QgkHUpPoocQw7zmExDzuWOO+517WnkTgPG9Mo7UaJ6aSftUH9UdgA0AtFDgAAAGwp3Ym/ft47957+aOmP9Lt3/a7+4It/oD/44h/od7f9rv5o6Y/0/qvvT2i0ANKuX5Eh6nm92nUXODZuRt6vWHz2+Fntvne3iktFlY6VdOCrB1T8SlH7vrRPu+7dpYVDC9qxuEPzhXltL25X9q6sXn705VBjm4Ygf2GepCRp0ALApJdR6u4vqeMMKkmfeUfUmKb9s+iWlnECSDaWqwIAAMCm1S/5N+h9SfqTn/8TffxfPlZuR077j+xXZi6jltuSvWarUq7oz37jz/Tj3/+xzvzfZ8Z+DwDSa+Nye53fB/Yc9gAAIABJREFUO7qX4Ot3XvfG4b32Jwq7Z8f50+eV35fXnvv3aO+De1VYKmjh0IJadku16zVl5jIyDEOe66llt+Rarpy6I+u2pdefeF0nv3cySjimjmTq5E17/4RpF4eSWFwBgM2IIgcAAAA2pShLUzl1R7933+/JnDF14BcOaH7/vLL5rOS337PWLOVv5VW/UdeNSzf0rz7/r/RbH/zWmO4AwGbQ79+hQQXYqNcI2v7ShUs6+OhB5ffntX1xuxYOLWjnwZ2yVi217JacuiO7Yis3n2u/8jll81nN7ZrTe6+9N7YixzgT0tNMtlNc+UQcRYCwm4hPs9CQhDEEkaZnNC0xBTBZFDkAAAAwcZ0CRJDlW4Kc169dWN898l1l7srowFcPaO8DezVfnFdmNiPXcmWtWqrfrCu3LaeZ7IwM09DalTX94dE/lJ4M3RUATMXyxWVlt2WVm88puy0rM2uqZbdkrVpyLVe+d2dWyYwhM2PKzJoyc6YyuUz7NZfRyrsrKjxUGGkcg5Kq/dbwP/RS782GgyZop50UnXb/nTGMGsco+l17lIR11A3ux2WS8RyXOOMV17PW69wkfJcAJAtFDgAAAExUHMu3bCx6dL8/aPmWQcu+/NXv/JUaHzd09y/drc8d/pz2/9f7tfPgTnmep8athmof1mRmTPm+L6/1yRIuaz9bk/69pG+MHhsAm8cDDzyg5eXl9d+LxaIkfepY3MfffPNNHT9+XJL6/vyjV38kGZJhGvI9X07NUe16TS27Jd/zZVdtuZYrz/Xke77k3/n31JSMmXab8jvlkYscowibPJ5msjnJf3U+jbiMWggIGs9J39ugwlxnPEl8BqTJPKNxfx5JjieA6aDIAQAAgIkbdfmWKMu6DHrfcz39xz/4j9p9727tWNyhnYd2as+De7T7nt1qfNSQaZrybE9uw5Vds2VXbeW2t5dxmd05K+evHRm/EG62CYDN6/nnn1e5XO75703Yf9fCHO/ey6Pvub7kOZ6cqqPGrYYycxk5dUeGYci1XDVvN+XUHLmWq5bTahc8Wn676BGTfgnKfn/5vbFNvyWPeiVSk7BMVdKSsVHiGGe/3ccGfeYbhS1wJCHuGwsdSTTuWMXxrPUaW9BnBsDWQZEDAAAAW97qT1dlVSzt371fsztmlcvnZM6Ychtue/bHjNFesiVrKjOb+eQ1l1H2rqx0Q/JX40v+AUi/Uqk08eMnTpwY/vOTJ/ST7/5EdtVWY7Wh2RuzMgxDdsWWMdPebNypOWrebsqu2XLqny52GKah0sO9xxSHIEnvuPdxGEeiN0mJ9n6SPLZuUeI56c88bSYZg7j7CFMcA7A1mNMeAAAAALaOXbt2qVarrb86rl27NtWfly8uyzANzWRnJENym67qH9VVKVfUuNWQU3fWl3LxfX99qZf1NetNUyqHjwcATFpxqSin7qzvM1RdqapSrqhSrqi6XFXtw5rqN+tqfNxQc60pu3qn0FF35VrtV+Ho9Jaq6idJSeskjSWsJI49iWNKuyTENAljALB5UOQAAADARPzKr/yKjh8/rr/7u79bf3X8xV/8xVR/rl2vtZekcr315F9tpaZKuaLa9Zqsj632OvUNVy279cla9d6dfT5kyKixXBWAdDj8+GFVyhXVb3y6yFFZrqi6UlXtxqcLHZ1ZHdaapSNPHJn28EO58mz/V/c5cQibuB22bE9SEsFRxznquKPc/6Q/80EGzSYZNaZRn5mkPFMAECeKHAAAAJiIe+65R0ePHtWDDz64/ur41V/91an+/Ln/6nPyWneWaFlrqn6jrspyRbfLt9cTftbHlpqVO+vUN1y5zfbyLb7ry/M9aX/4mADANJx+7bScurNe1KiUK6pcuzObY+XObI4bdTU+aqhxq13osNYsyZNOvnJyrGMLssRQv+PTTtqOOo7u+xvXXg6jxjHqOMPumZKUz3WYYfGUJhfTYe0mHdNRCzFhrwtg62JPDgAAAGx5pWMlteyWrNuW6h/VlbkrI6/lKTefk5kx5Xt+e9Pxqt3+i+bqJ+vUu7bb3pC3xJ4cANLjzFtndO6xc6pcrcipO8rms8rkMpIp+S1fLaclt+6uz+DwfV/PvP1MbP0PS1JGTX5Oy8ZxBfnr/e5jnTa92oaNRb9NnoO0HSTqOIe162WUeI4iakyjPpfjiumwzzxsTCf9rAVtl/QCGIDJYSYHAAAAtrzc9pwO/PwB1a7X1pdvqS5X19eoX1++5aO6rFWrXeiotAsddsWWPi8pO+27AIDgSg+X9NQPnlL1ZlWrP1391L4cndkctRs1VZYrcqqOnn7jaRWWxr8Xx6BlhAYdT3uyM85N1kfpa1h/Uce5FT+7znvTiGlSRP3cg9x7ku4TwPQxkwMAAACQ9A/+z3+g7x75rmrX2xuit5otZfPZ9c3IPddrz+ao2bJvt2d0NG831bJb0j+c8uABIILFRxb1XPM5XfjOBb3//feVmcu09xuSZJiGXMvVkSeOBF6iKkjScdTEZJyJzaRdK+w1RulzGm3D7qsxDlELB6Ned1ztJxXTpD8vAECRAwAAAJC0+97d+vo/+7r+w7/8D/JbvlzLVXZbVjO5GRmmIa/lqWXfWb6l+kmR49F/8ah+6P1QYrUqACl16tVTOvXqKa28u6LyO2VJ7ZkehaPjn7kBAAAwKoocAAAAwB2/+Du/qOxdWf3Vi38lu2ortz2nmdl2kUOe2mvUW+0ih2u5+sXf+UV97X/4mn74wg+nPXQAGFnhoYIKD1HYAAAA6UKRAwAAANjgG//zN3Tgawf0+q+8rspyRX7L10x2Rr58ebYnI2NobmFO3/m339Hnf+Hz0x4uAGAMwmyWzLI6AABMF0UOAAAAoMvPnfg5/dOf/VP97V/+rX7yxk/0d//P30mSPv+1z+vub96te755j2RMeZAAAAAAAIocAAAAQC+Gaeieb92je751z7SHAgCYMGZnAACQHua0BwAAAAAAAAAAABAFRQ4AAAAAAAAAAJBKFDkAAAAAAAAAAEAqUeQAAAAAAAAAAACpRJEDAAAAAAAAAACkEkUOAAAAAAAAAACQShQ5AAAAAAAAAABAKlHkAAAAAAAAAAAAqUSRAwAAAAAAAAAApBJFDgAAAAAAAAAAkEoUOQAAAAAAAAAAQCpR5AAAAAAAAAAAAKlEkQMAAAAAAAAAAKQSRQ4AAAAAAAAAAJBKFDkAAAAAAAAAAEAqUeQAAAAAAAAAAACpRJEDAAAAAAAAAACkEkUOAAAAAAAAAACQSplpDwAAAABAOhiGsf6z7/uRz+v3/qB2QfsGAAAAsLUwkwMAAADAUJ0iQ6fAsLHoEOY8wzDk+/76q9/xje0GvQcAAABga2MmBwAAAIBAOkWJYYWGfud1ihWD2sRl+eKyyhfLkqTSsZKKS8We5x16Kdr1rzw7vO2VZ8Ndc+P1hrXt7jtMX6O0DXvtsH30ah+kbdR7iqu/cfY5akz7XSvuZ2zU78OkPou4n9FJfPfG+Z0FAGAzoMgBAAAAYGLGvSTV+dPndenCJWW3ZSVD0p3LOHVHhx8/rNOvnY503SgOvTRa4nuU8+JuO+r1O+9FST4PimPUexo21rjHGbXPUWMa9FpRzotT3DEdRzyn8d2bxmcBAEAaUeQAAAAAMDG9lqga9l6QJaquvn1VZ4+fVX5vXgcfPajcfE6GachzPNlVW9aqpctvXNaLsy/qzFtnVHq4JGl4QjNIAnlQMjVMoSOMzjWjJEFHaRv02t2GxaPfDIOgcYx6T93XHPc4e/XZfd0g54ftM4q4YjpM1JhGbTdqPKfx3RvndxYAgM2APTkAAAAAJF6vfTw6rr59Va98+xUVl4q6+5t3a/GRRZWOlVQ4WtC+L+7T7vt2a+HuBe25f4/y+/M699g5XfvxtbGON8oSNkELKlET2qO0DXr9KO8NOi9IwSBqUj1MAnzYeUGXYup3XtjjQfoM03/3daexHFLYmEZpN0o8p/Hdm9ZnAQBA2lDkAAAAABBIZzbFsFkVQc/r1SbIed2FjrPHz2r3vbtVXCqqdKykA189oOJXitr3pX3ade8uLRxa0I7FHZovzGt7cbuyd2X18qMvBx7bJGzlRGbQAsCk/4q9u7+kjjOoJD5jUWOa9s8CAADEi+WqAAAAAAzVWTKqU4wYtrRUr/O6l50K+t6gvTrOnz6v/L689ty/R3sf3KvCUkELhxbUsluqXa8pM5eRYRjyXE8tuyXXcuXUHVm3Lb3+xOs6+b2TI8VlVCRhN7+we2eMq/9p9ZfE4goAANhcKHIAAAAACKTfZuDdxwdtGh7lvUFtLl24pIOPHlR+f17bF7dr4dCCdh7cKWvVUstuyak7siu2cvO59iufUzaf1dyuOb332ntjK3IESSxPO/k9SRRzPhFXESAtz1gSxhAEzygAAOlFkQMAAABAKi1fXFZ2W1a5+Zyy27Iys6ZadkvWqiXXcuV7d2aVzBgyM6bMrCkzZyqTy7RfcxmtvLuiwkOFkcYRZdPooO9vNkm43yvPtj+zXhtMjzPR3e/aoxYBkvSMTTKe45KEZxQAAIRDkQMAAADAQA888ICWl5fXfy8Wi5L0qWNxH/+bv/kbHT16VJL05ptv6vjx45/5+Uev/kgyJMM05Hu+nJqj2vWaWnZLvufLrtpyLVee68n3fMm/s/SVKRkz7Tbld8ojFzmiSGPyN6ok/yX/ND6HOAoBQWI66Xvrt5F7Zxy9ikpJkeRnFAAADEeRAwAAAEBfzz//vMrlcs8lo8IuLxXmePd+HX3P9SXP8eRUHTVuNZSZy8ipOzIMQ67lqnm7KafmyLVctZxWu+DR8ttFj5j0S4wOmy2wFRKqSb3XfhtTj3vD6kGFgKBFgDAFjiTEfWOhI4mSFCsAABANRQ4AAAAAA5VKpYkff+ihh9Z/PnHiRO+fnzyhn3z3J7KrthqrDc3emJVhGLIrtoyZ9mbjTs1R83ZTds2WU/90scMwDZUe7j2mOAxLXg9K/G6GxGsa7iHJY+slbEw3+zM2KmIAAMDmQJEDAAAAQCoVl4py6o6sVUv1m3VlZjPyXE+5+ZzMjCnf8+VaruyareZaU3b1TqGj7sq12q/C0ckvVbUVpDl5nNSxJ3VcaUU8AQDYPChyAAAAAEitw48f1uU3LrcLGzPtjcdz8zmZWVPypZbTklN3ZFfbhY7OrA5rzdKRJ45MZcxpWWYoqrD3EHTGy7RjEnWccSzXFCWmcV1rVIPufdSYRn1mkvJMAQCAeJjTHgAAAAAARHX6tdNy6o4qyxVVV6qqlCuqXKuoUm7/XvuwpvqNuhofNdS41VBzrSlrzZI86eQrJ8c6tiTvQzAuoyaPu2M2rhj2u27Q8UcdZ6/zNkNCflg8pcnFdFi7NMQTAACEw0wOAAAAAKl25q0zOvfYOVWuVuTUHWXzWWVyGcmU/JavltOSW3fXZ3D4vq9n3n4mtv6HJVXjTKYGSSb363OUtkFsvM6wv97vdazTplfbYQWAYcfD7onST9RxDmvXzygxjSpqTKMWpMYV02Gfedh4TuO7N+7vLAAAmwUzOQAAAACkWunhkp76wVOq3qxq9aer7dkc5Yqqy9X12Ry1GzVVlityqo6efuNpFZbGvxfHlWdJPoYxKJk9yb6G9Rd1nKP0mXSDxj+tmAIAgK2DmRwAAAAAUm/xkUU913xOF75zQe9//31l5jLyPV+SZJiGXMvVkSeOBF6iKkiSdJLJ9zj6HHfiN47rh73GtOIRte0k7y/qNSd1b9Pod7M/LwAAbFUUOQAAAABsGqdePaVTr57SyrsrKr9TltSe6VE4Ov6ZGwAAAAAmjyIHAAAAgE2n8FBBhYcobAAAAACbHUUOAAAAAMBnhNlImmV1AAAAMC1sPA4AAAAAAAAAAFKJmRwAAAAAgM9gdgYAAADSgJkcAAAAAAAAAAAglShyAAAAAAAAAACAVKLIAQAAAAAAAAAAUokiBwAAAAAAAAAASCWKHAAAAAAAAAAAIJUocgAAAAAAAAAAgFSiyAEAAAAAAAAAAFKJIgcAAAAAAAAAAEglihwAAAAAAAAAACCVKHIAAAAAAAAAAIBUosgBAAAAAAAAAABSiSIHAAAAAAAAAABIJYocAAAAAAAAAAAglShyAAAAAAAAAACAVKLIAQAAAAAAAAAAUokiBwAAAAAAAAAASCWKHAAAAAAAAAAAIJUocgAAAAAAAAAAgFTKTHsAAAAAwGZiGMb6z77vRzpvHO8BAAAAwGbETA4AAAAgJp0iQ6fAsLHoEPQ8wzDk+/76K473AAAAAGCzYiYHAAAAEKNO4WJYoaHfeVFnYERpt3xxWeWLZUlS6VhJxaViz/MOvRRpSLry7PC2V54Nd82N1xvWtrvvMH31GnfYsYa5dpjrRx1b1HjE1d84+xw1pv2uFfczNur3YVKfRdzP6CjfvXE/pwAAIP0ocgAAAAAJ06/oEVdB5Pzp87p04ZKy27KSIenOZZy6o8OPH9bp105Hum4Uh14aLYk5ynlh24YZa9TrS9GSz4PGFjUeo8Qiyjij9jlqTINeK8p5cYo7puOI57i+e1HbxfGdBQAAyUeRAwAAAEiYjcWLzjJU3T/3+n3Y8atvX9XZ42eV35vXwUcPKjefk2Ea8hxPdtWWtWrp8huX9eLsizrz1hmVHi5JGp7QDJJEHJRMHVcisnPNKMnT7vHEOdZh8QySfN74ftCxRY1H2FiMOs5efXZfN8j5YfuMIq6YDhM1plHbjRrPOL57435OAQDA5sGeHAAAAMAmMqjA8cq3X1Fxqai7v3m3Fh9ZVOlYSYWjBe374j7tvm+3Fu5e0J779yi/P69zj53TtR9fG+tYoyxhE7SgMsoyRWGSvmEFSQyHvUaQgkHUpPoosQg7zmGfcdjjQfoM03/3daeROA8b0yjtRonnKHGZ1nMKAADSjyIHAAAAEKPOclLDNv7ud94oG4b3K3BI0tnjZ7X73t0qLhVVOlbSga8eUPErRe370j7tuneXFg4taMfiDs0X5rW9uF3Zu7J6+dGXI49lHJKStJzmUkXDEv2THlt3f0kdZ1BJecY2ihrTtH8WcdoK9wgAwFbGclUAAABATDp7ZnQKFf2Wlhp03qB9Nwa916to0nn//Onzyu/La8/9e7T3wb0qLBW0cGhBLbul2vWaMnMZGYYhz/XUsltyLVdO3ZF129LrT7yuk987OXpwRkCCcvMLu3fGuPqfVn9JLK4AAACkBUUOAAAAIEb9ZlJ0Hx+0aXiU9wa1uXThkg4+elD5/XltX9yuhUML2nlwp6xVSy27JafuyK7Yys3n2q98Ttl8VnO75vTea++NrcgRJLE87eT3JFHM+URcRYC0PGNJGEMQPKMAACCJKHIAAAAAm9jyxWVlt2WVm88puy0rM2uqZbdkrVpyLVe+d2dWyYwhM2PKzJoyc6YyuUz7NZfRyrsrKjxUGGkcUTaNDvp+XK482x5nr42KJ5ncTUKie1qx6HftUYsASXnGevWVxsJBEp5RKTnfWQAAMF0UOQAAAIAYPPDAA1peXl7/vVgsStKnjg07fvnyZd1zzz2SpDfffFPHjx8f+ecfvfojyZAM05Dv+XJqjmrXa2rZLfmeL7tqy7Vcea4n3/Ml/86SV6ZkzLTblN8pj1zkiGKaScppLV+UlOTxRtP4HOJIWAeJ6aTvrd8G2Z1x9ErWJ0WSn1GJogYAAFsZRQ4AAABgRM8//7zK5XLPJaPCLC/Vvf9GHD+3D0ie48mpOmrcaigzl5FTd2QYhlzLVfN2U07NkWu5ajmtdsGj5beLHjHplxgd9lfYk06o9tuMeZybNCc1eTyNWGy8fvexfs9KL2EKHEmI+8ZCRxIlKVbdpvWcAgCA5KDIAQAAAMSgVCqNfPzee+9d//nEiRPx/PzkCf3kuz+RXbXVWG1o9sasDMOQXbFlzLQ3G3dqjpq3m7Jrtpz6p4sdhmmo9HDve4jDsOT1oATlOBOvk0rmJjl53JHksfUSNqbTesbSIi0xSPr4AADA+FDkAAAAADax4lJRTt2RtWqpfrOuzGxGnuspN5+TmTHle75cy5Vds9Vca8qu3il01F25VvtVODr5paqSKO5kb1qSx70kdexJHVdapT2eaR8/AAAIhiIHAAAAsMkdfvywLr9xuV3YmGlvPJ6bz8nMmpIvtZyWnLoju9oudHRmdVhrlo48cWQqY07LMkNRhb2HoDNeph2TqOOMY7mmKDGN61qjGnTvo8Y06jOTlGcKAABgGHPaAwAAAAAwXqdfOy2n7qiyXFF1papKuaLKtYoq5fbvtQ9rqt+oq/FRQ41bDTXXmrLWLMmTTr5ycqxjS9p6+f3GE2fCd9RrdY9xXDEcNRZRx9nrvM2QkB8WT2lyMR3WLg3x7JjEdxYAACQbMzkAAACALeDMW2d07rFzqlytyKk7yuazyuQykin5LV8tpyW37q7P4PB9X8+8/Uxs/Q9LqsaZiAySTB7U5zgLLxuvPeyv93sd67Tp1Tbs/QSJR5RYRB3nsHb9jBLTqKLGNOqzNa6YDvvMw8ZzHM/auJ5TAACweTCTAwAAANgCSg+X9NQPnlL1ZlWrP11tz+YoV1Rdrq7P5qjdqKmyXJFTdfT0G0+rsDT+vTiuPJusv7QelCxOwjgHjW+SfQ3rL+o4kx7/UQwa/7Riuhls5mcGAAAEw0wOAAAAYItYfGRRzzWf04XvXND7339fmbmMfM+XJBmmIddydeSJI4GXqAqSQJxk8j2uPseZGI3j2mGvMUqf02g7yfuLes1J3ds0+k3b8zJqWwAAkH4UOQAAAIAt5tSrp3Tq1VNaeXdF5XfKktozPQpHxz9zAwAAAADiRJEDAAAA2KIKDxVUeIjCBgAAAID0osgBAAAAABGF2fCYJXUAAACA+LHxOAAAAAAAAAAASCVmcgAAAABARMzOAAAAAKaLmRwAAAAAAAAAACCVKHIAAAAAAAAAAIBUosgBAAAAAAAAAABSiSIHAAAAAAAAAABIJYocAAAAAAAAAAAglShyAAAAAAAAAACAVKLIAQAAAAAAAAAAUokiBwAAAAAAAAAASCWKHAAAAAAAAAAAIJUocgAAAAAAAAAAgFSiyAEAAAAAAAAAAFKJIgcAAAAAAAAAAEglihwAAAAAAAAAACCVMtMeAACEZRjG+s++74c+b1j7oNcHAAAAAAAAMF3M5ACQKp0CRKf4sLEgEfQ83/f7Fi8Mw1h/3/f9vtcHAAAAAAAAMH3M5ACQOp0CxbAiRNDzerUJ6sP/9KF+9u9/pitvXZEkHXr0kA5+46D2f3l/qOsAAAAAAAAACI8iBwB0CbJc1Uf/30f63t//ntaurkm+lL0rK/nSB//2A8mUFg4u6Mk/fVILX1iY1LABAAAAAACALYciBwB06d6/o7vQ8e6fvKs//+/+XAuHFnTw6weVm8/JMA25TVd21Zb1saXaSk2//6Xf17f/+Nv68n/75UnfAgAAAAAAALAlUOQAgBDe+cN39O/+p3+nA187oN337tZ8YV7ZbVl5rie7YqvxcUP1m3Vl81llV7L689/6c3mON+1hAwAwVUFmSQ47L8g1ev1xQpD3AAAAAKQXRQ4AqdNJUgzbZyPoeb3a9FL/qK4f/PYPdODrB1T4ckF7v7hXOxZ3yMyYsqu26jfryl3PaSY3076O58tzPf3pr/+p9D9Kmg1zlwAAbA6d/w53/pvc77+1g87rbtPrGoP+ex/m/xYAAAAAkC4UOQCkysbER+f3jo0Jj2Hndf/cb5Pyje3+zX/zb7T989u16wu7tOf+PSocLWjXF3bJqTmqfliVmTHl+75aTkuu5cppOHJqjnKrOVnnLelX4o4GAADp0O+/s0HPGzYDY9AfNoT9o4fli8sqXyxLkkrHSiouFXued+ilQJf7jCvPDm975dlg1+p1nWFto7QJo9+9jfOeerWbdH/j6nPUeA66XthnJcqzNWr7cXwWcT+jYT6LOD77oO0BANhKKHIASJ1+iY7u40HPC/K+23D10zd/qkPfOKT8vrzy+/PaXtyubXu3ycpYsuu2spVse5mqbRte+axmd87K+ltLagW8QQAA8Bn9ih5xLVF1/vR5XbpwSdltWcmQdKeZU3d0+PHDOv3a6chjD+vQS9GStIPaDmsjjZ4sHbWPfu0HxSNqsSlKDIe1jftzi/szCxqrqDEdRdwxHcf3YJS4jPK8AQCA4ShyAEAA5YtlZeYyyuazysxlZBiG7Jqt+o26XNuV53qS306mmDOmzIypmexM+5WbkZEx5JdZBxwAgKh6FTbiKHBcffuqzh4/q/zevA4+elC5+ZwM05DneLKrtqxVS5ffuKwXZ1/UmbfOqPRwSdLwRGiQpOWgJGyQxGfQJO6wv94P0+cww+ISJPm88f2gY+u8FzYR3a+wMK5x9uqz+7rDzg3bX1RxxXSYqDGN2m7UmEaNS7++o8wsiaswCQDAZmFOewAAEEShUNA3vvENXb58ef3V8f3vf3/sP3/4nz6UaZiSIbWcluyqrdr1mirliuo36mquNeU2XLXslryWJ3lq/wWoIRmmIdM0ZaywHjgAAHHrLE/ZmenRvSxlv/ekdoHjlW+/ouJSUXd/824tPrKo0rGSCkcL2vfFfdp9324t3L2gPffvUX5/XuceO6drP7421vsJuqRPv/OCJGfD9hlUlL6HnRekYBA1qR4mAT7svHF8bnHEM2j/3deeRvI8bEyjtBslplHjMsp3FgAABEORA0Aq/Nqv/ZqOHTumPXv2rL86vvzlL4/959mds/I8T61mu8BRv1VXdbmqSrmi6odVNW41ZK1ZcmqO3IYr13bVclvyW758z2//JelcDIEAACCF+hUZgp7Xr53v+596dY4Ne6/j7PGz2n3vbhWXiiodK+nAVw+o+JWi9n1pn3bdu0sLhxa0Y3GH5gvz2l7cruxdWb386Msh7x7DBE0CT3oZpe7+kjrOoJKYTI8a07R/FqPYjPcEAMCoWK6ikqguAAAgAElEQVQKQGosLCz0PP7ggw+O/eeb/k35vq9mpSnrY0u1D2uSLzWrTZkzpjzXk1N31LzdlF215dQduZYrt+mq5bTk+Z5UinjjAACkWGdZqU6hot9+GoPOC7sReRDnT59Xfl9ee+7fo70P7lVhqaCFQwtq2S3VrtfWl6f0XE8tuyXXcuXUHVm3Lb3+xOs6+b2TI48hLJaoSadpf27TLg5t5ed1nJ/9Vo4rAADdKHIAQAB77t+jmcyMrI8t1W/WNZObkd/ylb2d1Ux2Rr7ny226sqt2u9BRseVU78zqaLiSKfm72ZMDALA19StKdB8fVLwIUtgI0/7ShUs6+OhB5ffntX1xuxYOLWjnwZ2yVi217JacuiO7Yis3n2u/8jll81nN7ZrTe6+9N7YiR5iEdNBk8pVn2+cO29tgnElT/vr8E3EUAcJuIj7NhHgSxhDEJJ5RCkAAAIwHRQ4ACMCYMbT0q0u6+K8vrm9I2rJbyubbRQ6pvVeH23BlV2xZty01K+1ZHc1KUzomiS05AABIhOWLy8puyyo3n1N2W1Zm1lTLbslateRarnzvzqySGUNmxpSZNWXmTGVymfZrLqOVd1dUeKgw0jgGJVWDbDrc73i//R06hY6w/cUpCUndQUWfcSa6o3xuw0Td4H5cJhnPcRlHvOL67NNSMAIAYNIocgBAQH/vX/49vffqe7p99bZ835drue0iR25Ghgx5LU9u05VTa//lp7VmqXGroW27t+n2t263NyIHAGALWV5e/tTvxWIx8PHZ2Vm99957On78uCTpzTffjO3n+f88LxmSYRryPV9OzVHtek0tuyXf82VXbbmWK8/15Hu+5N/ZF8Rs/+GD7/kqv1MeucgxqrQklJOcmJ1GzEb93ILGc9L3Nqi41hlPEp8BaXLPaFq+swAApA0bjwNACL/5/m/KMA3dvnJblXJFlWuV9v+uVFRdqar2YU31m3XVb9VVv1FXNp/Vb7z/G9MeNgAAE/fCCy/ohz/84Wc2/5Z6bwrefbzz+8b3Yv3ZlzzHk1N11LjVUHWlqkq5otqHNTU+aqh5uymn1t5jq+W02gWPlt8uesTkyrO9X9Lw5Ge/hHK/thuTuFH6iyqpBY6N9x7keJz99jsW5DMIW+BIQtyTMIZBplXg2HiMYgcAAKNhJgcAhHDXnrv0j//ff6w//sof6/a128rMZpTNZ2Wapnzfl+d6squ2PNfT9tJ2/aN3/pFmt89Oe9gAAEzFBx98oCeffPIzx0ulUs/zu4+fOHFiLD8v72jPGLGrthqrDc3emJVhGLIrtoyZ9mbjTs1p77NVs+XUP13sMExDpYd730Mchu2hEdawJG7c/QXtNwmSPLZuUeI5KHmehs9n3NIUgzSNFQCASaPIAQAhze2c02//l9/W2//72/rr3/trrf1sTbl8TpJk12ztOLBDX/0nX9XX/vuvTXmkAACgl+JSUU7dkbVqqX6zrsxsRp7rKTefk5kx5XvtZSntmq3mWnuPLafuyK27cq32q3B0uktVJV2aE7JJHHsSx5R2xBQAgM2DIgcARPT1f/51ff2ff11+y1f5YlmSVDpWkmGywzgAAEl3+PHDuvzG5XZhY6a98XhuPicza0q+1HJacuqO7Gq70NGZ1WGtWTryxJGpjHnj/gZJFjZ5PGwmSVKS0VHHOernFuX+g2xcP6l4Drr3UWMa9ZmZ5BJVcXxn0/C9BwBgmtiTAwBGZMwYWnxkUYuPLFLgAAAgJU6/dlpO3VFlubK+H0dnr631fbZu1NX4qKHGrYaaa01Za5bkSSdfOTnWsYXZm6HXsWGJ3Sj9BTVq8rh7LOPeK6Tf8bCbegcdZ9jPLSkFnmGCPFuTiumwdtOKaZTvbLekPwcAAEwLMzkAAAAAbEln3jqjc4+dU+VqRU7dUTafVSaXkUzJb/lqOS25dXd9Bofv+3rm7Wdi6z/q5uKddkGTwEHbjJpA3XjtsP0MG+OohZtBBYQwoo4zyuc2SjxHETWmUQtS44rpsM88bEyjxiXKZw8AAMJhJgcAAACALan0cElP/eApVW9WtfrT1fZsjnJF1eXq+myO2o2aKssVOVVHT7/xtApL49+L48qzgxPXg5K+cb83SYPGN8m+hvUXdZxJj/8ohj2v04hpkozy2adlNg8AANPETA4AAAAAW9biI4t6rvmcLnzngt7//vvKzGXke74kyTANuZarI08cCbxEVZBEZBzJyqjXGGeidBr3NUqf02gb174ao4haOBj1uuNqP6mYpuH+AADYqihyAAAAANjyTr16SqdePaWVd1dUfqcsqT3To3B0/DM3AAAAAERHkQMAAAAA7ig8VFDhIQobAAAAQFpQ5AAAAAAAfEaYTZJZUgcAAADTwsbjAAAAAAAAAAAglZjJAQAAAAD4DGZnAAAAIA2YyQEAAAAAAAAAAFKJIgcAAAAAAAAAAEglihwAAAAAAAAAACCVKHIAAAAAAAAAAIBUosgBAAAAAAAAAABSiSIHAAAAAAAAAABIJYocAAAAAAAAAAAglShyAAAAAAAAAACAVKLIAQAAAAAAAAAAUokiBwAAAAAAAAAASCWKHAAAAAAAAAAAIJUocgAAAAAAAAAAgFSiyAEAAAAAAAAAAFKJIgcAAAAAAAAAAEglihwAAAAAAAAAACCVKHIAAAAAAAAAAIBUosgBAAAAAAAAAABSiSIHAAAAAAAAAABIpcy0BwAAAAAASB7DMNZ/9n1/pPMMw/jMe4PaBe0bAAAAYCYHAAAAAOBTOkWGToFhY9Eh7Hn9jvm+v/7aeM6g9wAAAIBuzOQAAAAAAHxGp3AxrNAw6LxOwaL7+KDZGVFmbixfXFb5YlmSVDpWUnGp2PO8Qy+FvrQk6cqzw9teebb/e/3aDmozrH3QtlGvH6aPqOPrbjfp/sbV56jxHHS9YdcIG9NRnut+7cfxWcT9jEaNS5jPcNzfWwDAJyhyAAAAAABi12uJqjBtOwZd4/zp87p04ZKy27KSIenOqU7d0eHHD+v0a6cj9R/FoZd6JzAHJZE774VNlg/qL6xRxjeo/aDxRS02jRKPKOOM0ueo8QzTf5Tz4hR3TCf9HRp2zSj9xdEeABAeRQ4AAAAAQKzCFDh6nbvx917vX337qs4eP6v83rwOPnpQufmcDNOQ53iyq7asVUuX37isF2df1Jm3zqj0cEnS8ERokMTjoCRsvyTtsH6HJYX7JdLjKnREHV+/GQZh7ytsIjpsPEYdZ68+u6877Nyw/UUVV0yHiRrTqO3i+g4FjUvc34mg7QEA0bAnBwAAAAAgdoZhrL86v/c6J+xsj6tvX9Ur335FxaWi7v7m3Vp8ZFGlYyUVjha074v7tPu+3Vq4e0F77t+j/P68zj12Ttd+fC2We+onTIEi6HuDCi9xJkejjm/QeUEKBlGT6qPEI+w4hxW/wo4lznvude1pJM3DxjRKu1FiGiUu4/hOhG0PAAiHIgcAAAAA4DMGFSeGnbdx4/CNe3Z0t+tV4BjW39njZ7X73t0qLhVVOlbSga8eUPErRe370j7tuneXFg4taMfiDs0X5rW9uF3Zu7J6+dGXh99wSk1jmaKN/Q5L5k56fN39JXWcQSUxKR41pmn/LAAAycVyVQAAAACAT+lsFt4pOPRbPmrQeYP0K4xsvGb3cam9B0d+X1577t+jvQ/uVWGpoIVDC2rZLdWu15SZy8gwDHmup5bdkmu5cuqOrNuWXn/idZ383sko4cAWNO29E6ZdHEpicQUAgH6YyQEAAAAA+IzumRgbjwc5L2ibfn30On7pwiXtuneX8vvz2r64XQuHFrTz4E5t27tNcwtzmt0xq9x87pNXPqdsPqu5XXN677X3ooQhkHFspJ0ESR/fJB166dOvqNcIc940Cw2j3OckTbsY1G3YbJQkfLYAsBkxkwMAAAAAkHjLF5eV3ZZVbj6n7LaszKyplt2StWrJtVz53p1ZJTOGzIwpM2vKzJnK5DLt11xGK++uqPBQYaRxDEpyRk1c9tvboZNo7rfR9qQkISE7rXiMI1kddYP7cZn28xWHST+jw/btGFQkSsL3CQA2G4ocAAAAAIB1y8vLn/q9WCxGPn7r1i0dPnxYkvTmm2/q+PHjkX+e/8/zkiEZpiHf8+XUHNWu19SyW/I9X3bVlmu58lxPvudL/p3lsEzJmGm3Kb9THrnIEZcwSfJpJJ2T/Bfn04jHqIWAoPFMQgGrk6TvjCeJz4A0+Wc0yd8JANjqKHIAAAAAACRJL7zwgu677z6dOHHiM+/1W45q0PGN78Xysy95jien6qhxq6HMXEZO3ZFhGHItV83bTTk1R67lquW02gWPlt8uesSkX4Kz3yyDfucOulZ3X/32SxhXQjypydxpxSPsTJtuYQscSYj7xkJHEiW5wDHo3DD/TgAAgqPIAQAAAABY98EHH+jJJ5/8zPFSqdTz/EHHN763sXAS5eflHe0ZI3bVVmO1odkbszIMQ3bFljHT3mzcqTlq3m7Krtly6p8udhimodLDvccah6BJ7yjJ2UkmRJOUaO8nyWPrFiWeg4oLafh8xi2tBY7OcQodABA/ihwAAAAAgMQrLhXl1B1Zq5bqN+vKzGbkuZ5y8zmZGVO+58u1XNk1W821puzqnUJH3ZVrtV+Fo9NdqirO5Ow4Er1pTqAncexJHFPaJbnAAQCYHnPaAwAAAAAAIIjDjx9WpVxR/UZd1ZWqKuVK+7VcUXWlqtqNmuo362p83FBzrbk+q8Nas3TkiSNTHXvSk6Vhxzdsiaik3G/UcY467qgzdvq9us8Zt0GzSUaNadRnhgIHAKAfihwAAAAAgFQ4/dppOXVnvahRKVdUudYudFRXqqp9WFP9Rl2Njxpq3GoXOqw1S/Kkk6+cHOvYxrHE0KQKCKNer3uc494rpN/xsJt6Bx1nr/OG7bsQZDzTNiye0uRiOqxd2gocQWILAIgPy1UBAAAAAFLjzFtndO6xc6pcrcipO8rms8rkMpIp+S1fLaclt+6uz+DwfV/PvP1MbP0PS1J2J0U3nh/kr+PD9jeqUca3cXPqXm3D3lO/DcWDtB0k6jiHtetl1M87qqgxjfp8jSumwz7zsDGNEpdxficGtQUARMdMDgAAACABDMNYf0U5b1j7INcf1jeQBKWHS3rqB0+perOq1Z+uri9ZVV2urs/mqN2oqbJckVN19PQbT6uwNP69OMaxjNCgRHFSkqSDxjjJvob1F3WcafgMoho0/mnFdDMYFLvN8NwAQBIxkwMAAACYsk5xwff99UKE7/uhzuv8b69CRff1el2fAgfSZPGRRT3XfE4XvnNB73//fWXmMvK9O98B05BruTryxJHAS1QFSTpGTUyOmtAcd0I0juuHvcYofU6jbdh9NcYhauFg1OuOq/2kYhqlbVyfIcUMAJgcihwAAABAAmwsVgwqOAQ9L4xO0SPo9T6+/LHKF8uSpNKxknZ9YVcs4wDCOvXqKZ169ZRW3l1R+Z07z+TDJRWOjn/mBgAA/z979x5cx3neef7XfS4ACYAECEI8OKBAUaYoyTRjkiI1zoUhE9uRZ8ZKYpKKZSkTe6jEmVR2V9ndbFJ70WpVpUkmqcymnJ2J7dgjThKGssWlkkriXWU0iaVZLTO2xdAeKfRGDiNTpgCKVwDn1qev+0fjQCCIc+tzB76fqi41uvvt9+23GyrpffC+D4DuQJADAAAAWMXKzRpZzp/93J/pv/zhf5FMKRaPSZI8x5MC6f2ffL8++vmPtrKpQFmp3SmldhPYAAAAWI0IcgAAAAArXLlZGrUGOC6/fllf/P4vKrkmqdu+7zatGVkjwzTkOZ6crKPC9YL+9kt/q9e/9Lp+9ms/q433bGzFY3Slxf1aqS/LXVetfKXztdaN1aWeRNIspwMAAFYCghwAAADAKlBuYH25BOaLr73yt1f0+z/y+9r4no0avXdUg6lBJdYmFPiB7Jwta8ZS4WpBfev7lJnK6Nj+Y/rn/88/b/0DdYFO5lKpJc8KAAAAsBoQ5AAAAAC6QK15MerNn1GuvLT8zIClx76w7wvaeM9Gje8d18Z7NmooPaRYMiYnH87gyF/JK94flxEP2zN3cU5f2PsF6X+I1Lye06lcKlECGtNnpm/KpTK+Z7zhdqD7MDsDAACsNgQ5AAAAgA5b/Bf+pZ9LlgYlKl23dH+5WQL1DI4//9PPKzmU1MZ7N2rs3jGldqe0fst6+bav3NWc4mviMkxDvufLsz25liu34MrO2NKfSPqJOjsCLXHyyEmdO3VOibUJyZA0/wk4eUc7HtqhI88d6Wj7AAAAgEYQ5AAAAAC6QLngw9LjtV5X67lK17124jVNfv+kBlODGpoY0sjWEa3fsl7WDUu+68vNu3KyjuwhW8XBopKDSSUGE+of7pf9LZsgR4NqnfVRbqmqi6cv6tiBYxrYOKDJ/ZNKDibDoJTjy86GS42df/G8nu57WkdfOar0vnQrHgMAAABoKYIcAAAAAG7xzrfeUXxNXMl1SSUGEor3xeW5noqzRbm2q0CBDNOQETNkxk3FkrGFLd4fl+KScbnxZZlWu3KzdRYfKxfgOPHgCY3vGdfo9lENbBpYyKXiZB0VZgrKX80rOZhUZjqj4w8c16MvPNrSZwEAAABawex0AwAAAIDV7PLlyzdtS49fu3ZNX/va1xaOt2t/6tWpcGmsmBEOjOcd5a/klZnKqHCtIDtryy268j1fgR9IQTjgvhD4MEwFb6/8RNilwEMtuVRqua5S+VpyqJQcO3BMG7Zt0PiecaX3prX5A5s1ft+4xt47ppFtIxreMqx1E+vCWTrjQ0qsSeiZ/c9EahsAAADQSczkAAAAADrkN37jN7R9+3bt3bv3lnPFYlGSFI/HdePGjYXj7dr3HV+GES5t5OTCJOPx/ricnCMZklt0VZwrysk5couuPMeT74YBj8APZMiQ4a/smRydzKWyXNCkdM3JIyc1MDag0e2j2njPRqX2pDS8ZVie7Sl3ORcmijcM+e67uVScvCNrzpJ90pZI0QEAAIAeQpADAAAA6BDLsvStb31LH/vYx245d/vtty/sj4+PL+x/5CMfact+ak9KgR/IzoW5G3JXcpIh2VlbRmw++JF33g10FNww2GGHwQ4/8BWMr/yZHJ3KpVLp3LlT5zS5f1IDmwY0NDGk4S3DWj+5XtaMJc/25OQd2RlbycFkuA2ES5L1j/TLPmeXvS8AAADQjQhyAAAAALhFem9aTsGRNWMpfy2veH9cgRfIztgy46Z8z5dX9MIE1rOW7Kx9U7AjcAKJPNZtN31mWom1iTAJ/NqEzIQpz/ZkzVhyLTecZWO8m0vFTJgyk6biybjiyflcKpdW9gwcAAAArCzk5AAAAABwC8M0dPeP363MVEa5yzllL2WVmcpo7u258Ng7OeUu55S/mpd1w1oIdNg5W8XZovTeTj9Ba01PT9+0LT1+7do1vfzyywvH27U/dWZKMsL3F/iBnJyj3OWcMlMZ5a/mw1wqlruwtFgpl4pMLeRS0VTkbgEAAADajpkcAAAAAJb18J88rKfMp5S7nJMkuZarxEBCsWRMksKcDkVPds6WnQmXtbJnw4Tk+ilJK3S1qqeeekp33XWXDh48eMu5xXk4Fi8p1c59BWFOFSe7KJdK3pFhGHKtRblUrEW5VLwwlwoAAADQawhyAAAAACjrn/3FP9PJnzqpwAvkFt4NchimocALwhwPBUdO1lExU5TnevqZ//gz+v2Xf7/TTW+pN954Q4888sgtx9Ppd9foWhwEadd+em9Yv521VZgpqO9KnwzDkJ2Zz6Xihonki3NF2TlbTn5JsCMIFKQJdgAAAKB3EOQAAAAAUNadH75TP/XcT+kPfuwPZM1Y6h/uV6wvJtM0FQSBfMeXW3RlzVpy8o4+9dVPacuBLdLL1e+N5hvfMy4nP59L5Wpe8b64fNdXcjApM24q8AO5lruwrJidnQ905N0wZ4cbSKlOPwUAAABQO4IcAAAAACra+uGtejJ4Un/0kT/SP/zlPywMlkth7gff9fWeD79Hj/xft85sQPvteGiHzr94PgxsxMLE48nBpMyEKQWS53hy8o7sbBjoKM3qsGYtaUenWw8AAADUhyAHAAAAgJo8+sKjsrO2pr4xpe/99fckSbf/wO1K700rOZjscOtQcuS5I3q672llpjOSwlwqyYGkzKQpwwiDUq7lysmHy1YVZ4thgMOXdEQrNpcKAAAAViaCHAAAAABqlhxM6o4fuUN3/MgdHW4JKjn6ylEdf+C4MhczcvKOEgMJxZNxyVSYS8Xx5ObdhRkcQRDosdOP6fN/9vlONx0AAACoi9npBgAAAAAAmiu9L61HX3hU2atZzXx3RpmpjDJTGWWns8peyir3Tk65KzllpjNyso4++eInldpDMg4AAAD0HmZyAAAAAMAKNHH/hJ4oPqFTnzil17/8uuL98ZtyqbiWq50P79ShE4c63FIAAAAgOoIcAAAAALCCHX72sA4/e1iXzl7S1KtTksKZHqldzNwAAABA7yPIAQAAAACrQGp3SqndBDYAAACwshDkAAAAAADcwjCMhf0gCOq+rlL5xefqLQsAAAAsRuJxAAAAAMBNSkGGUoBhuaBEteuCICgboCidW+4awzBuOleubgAAAEBiJgcAAAAAYBml4EO1QEOt15VTCmo0YvrMtKbOzOcb2ZvW+J7xZa/b8plo97/wePWyFx6v7V7L3aeWslHL1aLcs7X7mbq5D+sp22h/VrpftXvU26eNftftehfN/kbreRfNamuUugEAtSHIAQAAAADoSSePnNS5U+eUWJuQDEnzsRIn72jHQzt05LkjbWvLls9EH/isVDZquVpVu78U7bmiPlMljfRFlHZGqbPR/qyn/ijXNVOz+7RcuUb6tJF+afXvHgCgeQhyAAAAAAA6YrlZHLXMCLl4+qKOHTimgY0Dmtw/qeRgUoZpyHd82Vlb1oyl8y+e19N9T+voK0eV3peWVH0gtJZBy0qDsLUMfNY7iNtouVpU65daBp8Xn6+1P0rn6n2OcoGFVrVzuTqX3rfatfXWF1Wz+rSaqH0atVyjfRq1X8rVHfV3FgDQGuTkAAAAAAB0lXL5OqQwwHHiwRMa3zOurR/aqon7J5Tem1ZqV0pj945pw10bNLx1WKPbRzWwaUDHHziut7/+dkvbW+uSPuWuixp8acYgai0Dw/Xeo5aAQdRB9XoGwKtd14r31oz+rLX+pffuxKB6vX0apVwjfRq1X9rxuwcAaB6CHAAAAACAW5RmU1SbVVHrdVHbsDTQcezAMW3YtkHje8aV3pvW5g9s1vh94xp775hGto1oeMuw1k2s02BqUEPjQ0qsSeiZ/c80vW2rXa2DwO1eRmlpfd3azlp142B61D7t9XcBAOheLFcFAAAAALhJacmoUuBicaBhceCh2nVL98vdZ7HF5ZaeP3nkpAbGBjS6fVQb79mo1J6UhrcMy7M95S7nFO+PyzAM+a4vz/bkWq6cvCNrztLzDz+vQ186FLlPooqSh6GRcmiOTvd/p4NDq/m7a9a7p08BoH0IcgAAAAAAbrFcAGK547VeF/X+i507dU6T+yc1sGlAQxNDGt4yrPWT62XNWPJsT07ekZ2xlRxMhttAUomBhPpH+vXac6+1LMhRz4B01IHPTgyY8hf172pG/9ebRLyTg+Ld0IZatOMbbdbvXq/0KQD0IoIcAAAAAICuN31mWom1CSUHk0qsTchMmPJsT9aMJddyFfjzs0pihsy4KTNhykyaiifj4dYf16Wzl5TanWqoHZUGVSsNXpYrV23gM2q5ZuqGQdkLj4fPvFyC6VYOdLei/6MmuG+VdvZnq7Sivxp59yuhTwGgl5CTAwAAAACw4O6779b09PTCVlL6+cyZMwvHXn755bbtv/TsS5IhGaahwA/k5BzlLueUmcoofzUvO2vLtVz5rq/AD6RgfukrUzJiYZmpV6eidElTlRIh15sQOWq5qLr5r85LwY7S1g6N9n+t/dnuwfBqydO7eXC+Xd9ove++l/sUAHoVQQ4AAAAAgCTpySef1IEDBxQEwcJWUu5Yu/bDA5Lv+HKyjgrXC8peyiozlVHunZwK1woqzhXl5By5livP8cKAhxeEQY8mWTrguXjgs9rgZaXBz3pniLQySXO3BjjKDTK3OvDTaP/XG+Dohn7vhjZU0s4AR7lj9f7udXufAkAvY7kqAAAAAMCCdDpd8fji8wcPHmzf/iMH9eZn35SdtVWYKajvSp8Mw5CdsWXEwmTjTs5Rca4oO2fLyd8c7DBMQ+l9yz9bM1RaTqmXdNNAeznd3LalovRnpcHzXng/rUYfAACWIsgBAAAAAOh643vG5eQdWTOW8lfzivfF5bu+koNJmXFTgR/ItVzZOVvF2aLs7HygI+/KtcIttauxfBwrXS8PHndj27uxTb2OPgUALIflqgAAAAAAPWHHQzvCHBxX8gtLVWWmMspMZ5S9lFXuSk75q3kVbhRUnC0uzOqwZi3tfHhnR9ocdTC23YO49Q4eV1uyp1sGo6O2s9F2R3n+ckuhLb5Hu3OylGtnpWuq9WnUb6aTS1Q1A7k4AKB1CHIAAAAAAHrCkeeOyMk7C0GNzFRGmbfDQEf2Ula5d3LKX8mrcK2gwvUw0GHNWpIvHTpxqKVtqyc3w3LHKg2sRi1Xq0bvtbR9rRrMbTSgErWd9fZ/twR4qqnWn1L7+rRauU71adR3X+l4t38XANCLWK4KAAAAANAzjr5yVMcfOK7MxYycvKPEQELxZFwypcAL5Dme3Ly7MIMjCAI9dvqxptUfNbl4qVw9AYCo5eqx+L5Rkp9Xal+1v8ivdryeQeRKorYzSv830p+NiNqnUb+rVvVptXdeb59G7ZdGfveYsQEA7cdMDgAAAABAz0jvS+vRFx5V9mpWM9+dWViyKjudXZjNkbuSU2Y6Iyfr6JMvflKpPa3PxVFtGaFKg76tKNculdrXzrqq1bbtwhkAACAASURBVBe1nd3e/42o9t11ok+7SZR332ifAgCiYSYHAAAAAKCnTNw/oSeKT+jUJ07p9S+/rnh/XIEfSJIM05Brudr58M6al6iqZeCxGYOT3ZifoxPP1UidnShbb16NVogaOGj0vq0q364+7cTzEcgAgPYjyAEAAAAA6EmHnz2sw88e1qWzlzT16pSkcKZHalfrZ24AAACgOxDkAAAAAAD0tNTulFK7CWwAAACsRgQ5AAAAAABYIepNbA4AANDrSDwOAAAAAAAAAAB6EjM5AAAAAABYIZidAQAAVhtmcgAAAAAAAAAAgJ7U1pkchmEs7AdBUPd11crXUq5a3QAAAAAAAAAAoDe0bSZHKdBQCjAsDTzUcl0QBGUDFIZhLJwPgmDZwEal8gAAAAAAAAAAoLe0dSZHKcCwXBAiynWtZGdsTb06JUlK70srOZjsSDsAAAAAAAAAAMDyVk3i8VqXynrpyZf0jc9+Q4UbBSXWJiRJbt5V/0i/7v/F+3XgyQMtbysAAAAAAAAAAKhuxQQ5Ks36WBrUKC1ttVh2Oqvf2/t78ixPg+lB3fa+22TGTHm2JyfnKH8lr9P/+2md/Xdn9ekzn9basbUtexYAAAAAAAAAAFDdiglySJWTjVeSncrqc7s/pzUjazT2/WMaTA2GszgCyc7ZKs4UtWbjGq25vEazF2f12Z2f1b/45r9oxSMAAAAAAAAAAIAatTXIUZpBUS0AUet11crX6nd3/q4Gxwc1cf+ENt69UUPpIcWSMTkFR9YNS7krOcXXxmXGTcmQ5r43p9/9vt+VfjFS8wAAAAAAAAAAQBO0LchRClqUAhdLZ10sTTZe7rql+6XzlXJuLA2WLD7/lV/4imRIt73vNo29d0ypXSmtn1wv3/WVv5pX7p2cjJghBZLv+PKKnlzL1cx3Z6T/W9JHIncJAAAAAAAAAABoQFtncpSbXbH0eK3XNXwukM4eO6vxPeMaSg9p3e3rtGHbBg3fMazCjYICP5BrubJztuysreRgcmHrH+mXc8aR8ZFos00AAAAAAAAAAEBjVlROjnpdOXdFhmmof32/koNJJfoTCvxAdtaW7/gyDENmzJQZNxVLxBTre3eL98clQ9LVTj8FAAAA0N0qzbqu5bqos7ZrrRcAAABA7zLbUcnmzZt1/fr1ha2k9PPMzIxee+21hePt2p96dUqGaYS5NgLJKTjKX80rM5VR4XpBds6WZ3sKvOCmZbEMcz74YZgK3uZ/lgAAAIByKi0zW+t1QRBUnbm93DXVygEAAADofW0JcoyNjWl2dnZhKyn9nMlk9J3vfGfheLv2nZwjwzDkOZ6cfJhkPHspq8zbGeXeyalwvaBipiin4MgrevIdX77nK/CDd3OHOCxXBQAAAFSyOP9eM65rpcK1gs7/h/M6/x/Oq3C90LF2AAAAAKhNW5arOnv2rH7iJ37iluNbt25d2L/99tsX9g8dOtSW/U27NinwAzk5R9aMpdyVnAzTkJ2xZcQM+Y4vO2erOFuUnbNvDna4vnzfV5DiL8MAAACATmrGslR/+at/qVe/8KrsnK1Ef0KS5BZcJQYT2vvze/XBX/9gU9oKAAAAoLlWdU6O9H1puUVX1qyl/NW84v1xBV6g5GBSZtxcSDxezBTDQEfWlpNzFoIdgRtI6U4/BQAAALB6LZejo55Ax9xbc/r83s9LvjQ8Oaw1o2tu+oOnwtWCvvG739A3//039elXP62hiaFmPwIAAACABqzqIEesL6atP7pVl85eWghseLYX7ifCPB2eHS5lZWfCGR3FbFFOzpE9Z0vb1KYFvwAAAAA028x3Z/TFf/RFDYwNaGzHmAZTg0qsTYSzvfPhbO/ChoL6r/Rr7u05fX7P5/Vz3/i5TjcbAAAAwCKrOsghSR9//uP6V8P/SrnLOUmSa7kLQQ7DMOS7vlzLlZ2zZc/NBzoyRdl5W/p4hxsPAAAA9IDS7IpyScfrva5ZPrfrcxqeHFZ6X1ob79mowfFBxZIxuQVXhRsF5a/klehPhH8AZUhz35vT53d/Xvpv2tI8AAAAADVY9UGO5GBSh/7gkP78F/5cgRfILbhKDiQVS8YkUwq8IJzNUXBkZ20V54pyC66OPHtEJ799UiIlBwAAAFBWKWhRClwsXkpq8dJS1a5bul86vzQgUmu5P/3ZP1UsHtPYjjGNvXdMqV0prZ9cL8/1VLhaUOJSQoZpKPADeY4n13LlWq5mvjsj/bmkf9pYvwAAAABojlUf5JCk9z3yPhkxQ3/8M3+s4lxRfev7FE/GZcTC/6nxHV9u0VVxtijHcvTQlx7SPYfukZ7qdMsBAACA7lcuR8bS47Ve14xz3/z339TE/RMaHB/Uus3rNPKeEQ3fMSzrhhUuV1VwwtncWVvJoaSSg0klB5JaM7xGzt84qyLIUWtC90rXRT1XT/0AAABY3QhyzNvx8R3acmCLTvzTE7r6/12V7/oyY2HCDd/zZcZNjd07pkdfeFRrN67tcGsBAAAARHX59cuKJ+PqX9+v5GBS8f64Aj+QnbHlOZ4MGTJjpsy4qVgiplgypljf/NYfC/8Y6srKHnRfPPOlNMOmUiBiueuWlqn13HI/AwAAAOUQ5FhkMDWoT5/5tK7//XVNfX1K3/1P35Uk3XHgDk3cP6GR94x0toEAAABAD7n99tt17dq1hZ9HR0clSTdu3NBbb72l97///ZKkb33rW23d1zclmZIRM6RAcgqO8lfy8h1fvu/LztpybVeBF9w0KG+YYfDDMAwZU+3JG9JJS5cSq/e6qEGKKAGO6TPTmjozJUlK701rfM/4stdt+UykJunC49XLXni8vnsuvl+5suXqrLeuetoSpZ7lytdSdmm5dtfXqjqb/d5q+VbK1V3v9UtFKd+Kd9HsbzTKu6jnPTSrTgBAbQhyLGPDtg3asG2D3vfI+zrdFAAAAKAn/dIv/ZLeeecdZTKZhWOlIEcul9Obb765EHho9/5EfkKGDPmuLztnq3CjoMSahOy8LcMw5Fqu7DlbTt6RV/TC4IfnK/DDoIchQ4HNLINGVAuI1BosOXnkpM6dOqfE2oRkaCFnopN3tOOhHTry3JGmtruSLZ9pbMC8nmtK55oxaNpoPeXKV+qPqMGmam1tdjuj1Nns91ZrX0Xt00Y0u0/LlWukT5vVL/XcpxPvAgBWO4IcAAAAAJpu/fr1Wr9+/bLnNm/erM2bNy/8/JM/+ZNt3b/41xcVBIGcnCNrxlL+cl6GYSiZScqMm/IcT07OUXGuKDtryyk48ixPvu3Ld315gScjvfJncjRTvctTVVu66uLpizp24JgGNg5ocv+kkoNJGaYh3wln4lgzls6/eF5P9z2to68cVXpfWlL1gdBaBp8rDcLWE+ioRbX2Nqu+qPWU+8v2WttXOlfvoHC5wEKr2rlcnUvvW+3aeuuLqll9Wk3UPo1artE+jdovjehEnQCwWpmdbgAAAAAAtFN6b1qOFQY4CtcKyr6TVWYqs7Dl3skpfzWvwo1CGOjI2LLzthwrDHbIkYI0Mzlq1ez8GhdPX9SJB09ofM+4tn5oqybun1B6b1qpXSmN3TumDXdt0PDWYY1uH9XApgEdf+C43v76202rfzlR/jI/yqB6lPqqaUY9S6+r5dmiDqrXMwBe7bpal2Iqd129bWnmMy93704sh1Rvn0Yp10ifNqNf6p2B06l3AQCrGUEOAAAAAKuKmTC17ce2KTudVe5yTrlLOWWnwkBHdjqr7KXweOFa4aZAR2l2h7YrXBpphSstGVUpH0e161qRQPzYgWPasG2DxveMK703rc0f2Kzx+8Y19t4xjWwb0fCWYa2bWKfB1KCGxoeUWJPQM/ufaWobGrUSBkBrDQC0+6/Yl9bXre2sVTd+K1H7tJffRTe+BwDAu1iuCgAAAMCq8/E//rh+beDXlLuckyS5lqvEYEKxREwyJN/15VpuGNjIFFWcLYbBjrwtPdzhxrdBKWdGKXBRbvmoatct/ufi85VyclQ6d/LISQ2MDWh0+6g23rNRqT0pDW8Zlmd7yl3OKd4fl2GE+VY82wvfYd6RNWfp+Yef16EvHWq8cxrQjYO3WF4z8540Un+n6mNQP8TvLAD0BmZyAAAAAFh14v1xPfSlh8LlqqYXLVc1Hc7myF3KKXd5ftmqawUVZgpyLVcff/7jq+b/ooIgWNiWHq/1uqVbLeUqnTt36pxGto1oYNOAhiaGNLxlWOsn12vtxrXqH+5X37o+JQeT724DSSUGEuof6ddrz73WSHdUVE8i8VYnC28mBnjfteUzN29R71HPdZ0MNDTynO3UyjZ2w3sAANSGmRwAAAAAVqV7H7pXRszQyYdPqjhXVN/6PsX6YjJMQ/Ilzw1nAhTninIKjj5+6uO6+8fvlr7V6ZavTtNnppVYm1ByMKnE2oTMhCnP9mTNWHItV4E/P6skZsiMmzITpsykqXgyHm79cV06e0mp3amG2lFpULWRPBtRtGvwtRsGeS88/u7Ae7nE461Q7t6NDIBHTXDfKu3sz1ZpdSJ3AEB3WyV/gwQAAAAAt7rn0D365Xd+WaN3jyp7KauZ785o5s0Z3bhwQ3Pfm1PhWkG37bxNv3rtV8MAxyowPT1901bL8dOnTy/8/PLLL7dkf+rMlGRIhmko8AM5OUe5yzllpjLKX83LztpyLVe+6yvwAymYXyrLlIxYWGbq1alGuiayZg4at+uvy7v5r9ibMauiXqVk0lGTStfan+0OMFRLnt7NAY9WfqPd/NwAgFsxkwMAAADAqrZmZI0e++vHNHdxTm9/7W1d+E8XJElbDmzRxP0TWrd5XYdb2D5PPfWU7rrrLh08ePCWc+USiC9dVqql+4HkO76crKPC9YLi/XE5eUeGYbw76ybnyLVceY4XBjy8IAx6NEm5AdVqswyauUzVag1wlEtM3eqE1eUCAeXe+VL1Bji6od9Lz9et2hHg6Ib3AACoDUEOAAAAAJC0bvM6rdu8TvcevrfTTemoN954Q4888sgtx9Pp9LLXp9Ppm84tDpA0c396XTh7xM7aKswU1HelT4ZhyM7YMmJhsnEn54QJ4nO2nPzNwQ7DNJTet/wzNEO1Qe9KA8a1DKqu9gDHYt3ctqWi9Gej38pK1+7fhU62AQBQG4IcAAAAAICuN75nXE7ekTVjKX81r3hfXL7rKzmYlBk3FfiBXMuVnbNVnC3Kzs4HOvKuXCvcUrsay8fRKQQ4quvGtndjm3odfQoAWA5BDgAAAABAT9jx0A6df/F8GNiIhYnHk4NJmQlTCiTP8eTkHdnZMNBRmtVhzVra+fDOjrS50dkZ3RrgqHXmSqcHo6O2s9HlmqI8fzd8B0vrW06jfRr1m2lXH3TTewAA1IbE4wAAAACAnnDkuSNy8o4y0xllL2WVmcoo83ZGmanw59w7OeWv5FW4VlDhekHF2aKsWUvypUMnDrW0ba3IX9CtAY5y5cv93Czl7hs1qXet7Vzuukp19spAeLX+lNrXp9XK9UqfAgA6g5kcAAAAAICecfSVozr+wHFlLmbk5B0lBhKKJ+OSKQVeIM/x5ObdhRkcQRDosdOPNa3+aoOxzRqEXVxPtb+q71Q9i2c7LFe22l/kVzteb26TcqK2s1q55bTrvVWqt9LxpfVGDUi1qk+rvfN6+7SRby2qTtQJAKsdMzkAAAAAAD0jvS+tR194VNmrWc18dyaczTGVUXY6uzCbI3clp8x0Rk7W0Sdf/KRSe1qfi+PC46tz0LLSYHY766pWX9R2NlJnt6vU/k71KQAAUTCTAwAAAADQUybun9ATxSd06hOn9PqXX1e8P67ADyRJhmnItVztfHhnzUtU1TK42s5B+1bV16p66r1HI3V2omyz8mo0ImrgoNH7tqp8u/q02e+jU/+uAABURpADAAAAANCTDj97WIefPaxLZy9p6tUpSeFMj9Su1s/cAAAAQHcgyAEAAAAA6Gmp3SmldhPYAAAAWI0IcgAAAAAAsELUk0iaZXUAAMBKQOJxAAAAAAAAAADQk5jJAQAAAADACsHsDAAAsNowkwMAAAAAAAAAAPQkghwAAAAAAAAAAKAnEeQAAAAAAAAAAAA9iSAHAAAAAAAAAADoSQQ5AAAAAAAAAABATyLIAQAAAAAAAAAAehJBDgAAAAAAAAAA0JMIcgAAAAAAAAAAgJ5EkAMAAAAAAAAAAPQkghwAAAAAAAAAAKAnEeQAAAAAAAAAAAA9iSAHAAAAAAAAAADoSQQ5AAAAAAAAAABATyLIAQAAAAAAAAAAehJBDgAAAAAAAAAA0JMIcgAAAAAAAAAAgJ5EkAMAAAAAAAAAAPQkghwAAAAAAAAAAKAnxTvdAAAAAAAA0ByGYSzsB0FQ93W1lF/umsXHqtUNAADQTMzkAAAAAABgBSgFGsoFHmq5LgiCqsGR0jVLryt3HAAAoJWYyQEAAAAAwApRCjAEQVA2yFHPdYuVAhzNMn1mWlNnpiRJ6b1pje8ZX/a6LZ+Jdv8Lj1cve+Hx+u65+H7Vyi6tu9666mlLlHqWK19L2ajP1az6WlVno/1Z6X7N/lYa/a7b9S6a/Y1GeRf1vIfl6oxaL4D2IsgBAAAAAABqUmk5q1qXyjp55KTOnTqnxNqEZEiav9TJO9rx0A4dee5IU9tcyZbPNDZg3sh1jahUR+lclMHnSv0R9bmqtbXZ7YxSZ6P9WU/9Ua5rpmb3ablyjfRps/ql3vtE7RsAnUeQAwAAAAAA1GRp/o7FM0IWW27Wx8XTF3XswDENbBzQ5P5JJQeTMkxDvuPLztqyZiydf/G8nu57WkdfOar0vrSk6gOhtQw+VhqEbdUAZumerRrIrtYvtQw+Lz5fa39Efa5ygYVWtXO5Opfet9q19dYXVbP6tJqofRq1XKN92urfoeXatLjexccJdADdjZwcAAAAABCRYRgLW5TrqpVv9DzQLS6evqgTD57Q+J5xbf3QVk3cP6H03rRSu1Iau3dMG+7aoOGtwxrdPqqBTQM6/sBxvf31t1vapih/mV9rQKXVg6G1DAzXe49aAgZRB9XrGQCvdl2tSzGVu67etjTzmZe7dycGzuvt0yjlGunTZvRLlBk4y11PYAPoDQQ5AAAAACCCVid5XprgebkACYmesVTpO6kl8FbLdc1y7MAxbdi2QeN7xpXem9bmD2zW+H3jGnvvmEa2jWh4y7DWTazTYGpQQ+NDSqxJ6Jn9z7SlbbVaCYOdtQYA2r2M0tL6urWdterGbyVqn/byu6h3KbpefEYAIZarAgAAAICIWpnkuZKoCaCvvXFNkjS6fbThNqD7lL6vpYE16dalpSpdt3S/3PdbrtzScyePnNTA2IBGt49q4z0bldqT0vCWYXm2p9zlnOL9cRmGId/15dmeXMuVk3dkzVl6/uHndehLhxrrmAYxsNk7ov71frPr71R93Rhc6QR+Z4HVhyAHAAAAAHShSgPKUu1Jns9+4az++rf/Wtf+/priyfB/AV3b1ei2Uf3AL/+Adh3d1eSWo5PKfQtLj9d6XdT7L3bu1DlN7p/UwKYBDU0MaXjLsNZPrpc1Y8mzPTl5R3bGVnIwGW4DSSUGEuof6ddrz73WsiBHLQOhnR40j4IB3nc1IwhQbxLxTn4r3dCGWrTyG+2VPgDQXAQ5AAAAAKALLZ2tsfTnSuckyS26+uI/+qJmvzerNSNrNLF3QmbSXEjynLuS01/8d3+hr//br+vnvv5zMmLk9UDzTZ+ZVmJtQsnBpBJrEzITpjzbkzVjybVcBf78rJKYITNuykyYMpOm4sl4uPXHdensJaV2pxpqR6VB1SjJq3tBN7T7wuNh3y+XtLkdA93ljkfpm6gJ7lulnf3ZKq1O5A5g9SAnBwAAAACsMMVMUb+z9XdUnClqyw9t0Z0fvFMTH5hQaldKt73vNm28Z6Nue+9tWn/Het1484Z+e/K3ZefsTjcbDZienr5pW3w8k8no5ZdfXjjWzv2Xnn1JMiTDNBT4gZyco9zlnDJTGeWv5mVnbbmWK9/1FfiBFMzPUjIlIxaWmXp1KkqXNKwXB427+a/YS8GO0tYOpQTWURNZ19qf7f5WqiVP7+Zvt5XfaDc/N4DWYiYHAAAAAERUmkFRS5LnZuXjqMVn3/dZJQYS2vyBzRq9e1RD6SHFkjG5litrxlL+aj78q/qkKcMwNPu9WX1u5+ekT7aleWiyp556SnfddZcOHjx4y7nlktO3cz88IPmOLyfrqHC9oHh/XE7ekWEYci1XxbminJwj13LlOV4Y8PCCMOjRJOUGVKvNMujGYEE53drmckmbW53MuVwgoNw7X6reAEc39Hvp+bpVOwIc3fAeALQfQQ4AAAAAiKCTSZ4rnfur/+WvVJwr6s4P3amxHWNKvT+ldbevU+AHKlwrKHs5KzNuhgPPpUTPRVezF2allyQdaLRn0AlvvPGGHnnkkVuOp9NpSbopANLW/UcO6s3Pvik7a6swU1DflT4ZhiE7Y8uIhcnGnZyj4lxRds6Wk7852GGYhtL70tUeP7Jqg96VBoy7aVC1m9pSTje3bako/dkr30qntKsPeA/A6kSQAwAAAAAi6lSS53LnfNfXq597VaPbRxcSPI/ePaqRO0dUuF6QYRphUCPvysmFyZ6LmaKSg0n1re+T83VHxg+TmwPNM75nXE7eWZhBFO+Ly3d9JQeTMuOmAj+Qa7myc7aKs0XZ2flAR96Va4Vbaldj+ThWul4euO3Gtndjm3odfQqg1QhyAAAAAMAKceMfbsjNu+of6VffUJ8SaxMyDENO3gkTPJtGmNg5YSqWjCnWF1O8L654X1yJNQnpqhTcaN4SQYAk7Xhoh86/eD4MbMTCxOPJwaTMRDijyHM8OXlHdjYMdJRmdVizlnY+vLMjba40GNtNA7b1tqXWmSudfrao7Wx0uaYoz99N30qlZ2+0T6N+M+3qg0beQ6/8XgAoj8TjAAAAAFCn0dFRZTKZha2k9LNt27pw4cLC8XbtT5+ZlgwplohJktyiq/y1vDJTGRWuFeTkHHm2p8Cfz9EwnxDaiBky46ZMw5Q6k+MZK9iR547IyTvKTGeUvZRVZiqjzNsZZabCn3Pv5JS/klfhWkGF6wUVZ4uyZi3Jlw6dONTStnVz/oJqGh14XfrsreqLcveNmtS71nYud12lOntlILtaf0rt69Nq5XqlTxdr1+8FgOZiJgcAAAAA1OFTn/qUZmdnNT09vXBsaGhIkhaObdiwQV/96lf1qU99SpLatp+/mpdhzuc5KDiybljKXsouJHl2CuESVW7BlWd78h1fvudLfrj8lSlTft5vep8BR185quMPHFfmYkZO3lFiIKF4Mi6ZUuAF8pxwGbXSDI4gCPTY6ceaVn+1gcpmDsLWMgjdaJ2L71Xtr/eXO1Yqs1zZan+RX+14vblNyonazmrlltNIfzYiap9GHXhvVZ9We+f19mk7foeWa0eUvgHQHQhyAAAAAEAdtmzZUvbc9u3bF/ZLAYh27m/6vk0K/CDMbzBTVO5KToZpyM7aC8sE2TlbxbminNx8gmfbk+d4CtxAXuAp2MRyVWi+9L60Hn3hUT2z/xlZc5b6R/oVT8ZlxAwFfiDf9eVa7sIMjsdOP6bUntbn4litA5fllnVqRX80UlfUsu18vnartCQXfdqY1fCMwEpFkAMAAAAAVojx+8bl2Z6Ks0Xlr+UVXxNX4AdKZuaTPHthkudiZj7vQSnJs+XKLboKvEDGBInH0RoT90/oieITOvWJU3r9y68r3h9+n1K4bJprudr58M6al6iqdUC32WoZDG61ZtRR7z0aqbMTZZuVV6MRrfpWGm1vt/dps99HN3wLAFqLIAcAAAAArBDJwaRu//7bde0719Q31CczbspzPCUHFiV5tsMkz8VMUcXZouyMLTtny87a0qQUxJnJgdY6/OxhHX72sC6dvaSpV8MkMOl9aaV2tX7mBgAAWHkIcgAAAADACvKx4x/Tv9n+b5S9nJWMMPl4ciCpWDJMRl5aFqi0bFUp0OFarnS4w43HqpLanVJqN4ENAADQGIIcAAAAALCCrJ9crx9+8of1yq+9osAP5BScd4McxnySZ9sLk5Bn5wMdmaJ+9F/+qF7MvSitgokchvHuklxBUP6By11XrXyj57H61JNImuV0AAC4GUEOAAAAAFhh9v+P+5XoT+iv/ue/kp2x1TfUp1hfTIZpKAgC+c78bI6MLcdy9GO/+WPa91/t04tPvdjpprdcKcAQBIEMw5BhGBUDEctdV/rn4mBFrfcvVx8AAACiIcgBAAAAACvQB/7bD2jyByd18uMnlb2Uled6iiXCJas8O9wf2Dign/mPP6PUntW1ZNDiYMVygYp6r6u1XJQAx/SZaU2dmc9bsTet8T3jdZVHb2B2BgAA0RHkAAAAAIAVKn1/Wo//w+N66/99S2/+5Zv63unvSZJu/8HbtfVHt2ryhyY73MLVp9alqk4eOalzp84psTYhGVpYRszJO9rx0A4dee5Ii1sKAADQGwhyAAAAAMBKZkiTPzRJQKNLVFu66uLpizp24JgGNg5ocv+kkoNJGaYh3/FlZ21ZM5bOv3heT/c9raOvHFV6X7rdjwAAANBVCHIAAAAAANAFLp6+qBMPntD4nnGNbh/VwKYBJdYmwgTyWUeFmYLyV/NKDiaVmc7o+APH9egLj3a62QAAAB1FkAMAAABdpdalXCpd1+5zAHpLaQZFtTwbtV7XrHLHDhzT+J5xje8Z18Z7NmpoYkhmwpSTc1S4XlDflT7F++IyY6YkKXMxo2f2PyP9T3VVAwAAsKIQ5AAAAEDXKA0IlgYHyyXprXRdtXPlloqJeg5Ab1n874bSzyWLf7erXbd0v5ZySwMfi8+dPHJSA2MDGt0+qo33bFRqT0rDW4bl2Z5yl3OK98dlGIZ815dne3ItV07ekTVnyT5pS6ToAAAAqxRBDgAAAHSVpQOFUa6r9R71tqlWuXdyevsbb+utV96SJE3uuw/n2gAAIABJREFUn9TE3gkNbBpouC0AGlfud3rp8Vqvq+d8uXPnTp3T5P5JDWwa0NDEkIa3DGv95HpZM5Y825OTd2RnbCUHk+E2kFRiIKH+kX7Z5+yK7QEAAFjJCHIAAAAANahluapipqg/+id/pKlvTMmMmYolYgqCQF//na/L931N3D+hn37hp5VYm2hXswH0gOkz00qsTSg5mFRibUJmwpRne7JmLLmWq8Cfnx0SM2TGTZkJU2bSVDwZVzwZl+KScanxgC4AAEAvMjvdAAAAAKBdFi8js9zsj3LnSudL23Ln/+5P/k6/ueE3lbmY0eQPTurOD92pO37kDk3+4KTSe9PauH2jrn77qn593a/rO1/5TqseEUAF09PTN22S9PLLLy+c79T+S8++JBmSYRphkvGco9zlnDJTGeWv5mVnbbmWK9/1FfiBFMwHXk2FgQ/DlKbq7g4AAIAVgZkcAAAAWFXKratf7Vwl3/7jb+tPH/tTbf7+zdqwbYMGU4NKrEnI93zZWVvWDUv5q3klBhLKvZPT848+r4/94ccafxgANXvqqad011136eDBgzcdX5ozoxP74QHJd3w52TDJeLw/LifvyDAMuZar4lxRTs6Ra7nyHC8MeHhBGPQAAABYxQhyAAAAoKuUEv9WCzJUuq6We1RKHl5PonHf9XXy8End/oO3K/X+lDbeu1FDE0OKxWOyc7YK1wrKXc4plozJMI1wINPz9eyPPyv9bxUfEUCTvfHGG3rkkUduOrY46NGx/UcO6s3Pvik7a6swU1DflT4ZhiE7Y8uIhcnGnZyj4lxRds6Wk18S7AgCBWmCHQAAYHUiyAEAAICusXjJqNLPJYsDDZWuq3aPxdctVunc0oDJ4vNf+okvaWjzkEbuHNHGezZqfPe4hu8YlmM5yr0TBjeCIJDv+vJsT67lysk7Kg4VZT1rSQ/X2UkAVpzxPeNy8o6smXDWV7wvLt/1lRxMyoybCvxAruXKztkqzhZlZ+cDHXk3zNnhBlKq008BAADQGQQ5AAAA0FXKzZhYLvDQ6D1qPVfuvO/6+vsX/l6TPzSpgU0DGkwNamjzkAbHB8OEwQVXdtYOkwkPJJRYO78NJNQ30ifr7ywZIlkwAGnHQzt0/sXzYWAjFiYeTw4mZSZMKZA8x5OTd2Rnw0BHaVaHNWtJOzrdegAAgM4hyAEAAABENH1mWvH+uJIDSSXWJGTEDLkFV4VrBblFV4EXLCQTNmOmzISpWCKmWDLcjIQh/22/048BoAscee6Inu57WpnpjCTJtVwlB5Iyk6YMI1yyamEm2FxRxdliGODwJR2RxGpVAABglTI73QAAAAAgl8vp/PnzeuuttxY2SfrKV76ycE037k+fnQ7/i9rUwpr5ucs5ZaYyyl/Nq5gpyi248h0/TA4cSDL0buDDMGVMM5MDQOjoK0flZB1lLmaUmcpobmpO2amsMtMZZS9llbuSU/5KXoXrBeWv5xUEgR47/Vinmw0AANBRzOQAAABAx/3Wb/2WJicn9dGPfvSm45s2berq/fjluORLXtGTnbWVv5ZXrC8mJ+fIMA25lqtiZj5RsOXIs70w4OEFCvxAgQIFcf78GkAovS+tR194VM/sf0bWnKX+kX7Fk3EZMUOBHyzM5ijN4Hjs9GNK7UlJf9bplgMAAHQOQQ4AAAB0hQsXLmhsbOymY3v37u3q/ct9lxUEgeysrcKNgpKXk5Ike8iWEQuXl3HyTpgoODOfKLjoLgQ7/MCXMcFMDgDvmrh/Qk8Un9CpT5zS619+XfH+eDgTTFoInu58eKcOnTjU4ZYCAAB0B4IcAAAAQES37bxNXtGTdcNS4WpB8b64Ai9Qca6oWCKmwA/kWq7snL0Q6LBztpyCI7fgSp4UjDGTA8CtDj97WIefPaxLZy9p6tUpSeFMj9SuVIdbBgAA0F0IcgAAAAAN2PWpXTp38pySQ0kZMUOe7SkxkFAsEZMkeY4nJ+/IzoaBjuJcUXbWlpWxpN0dbjyArpfanVJqN4ENAACAcghyAAAAAA348X/34zr3f57T3NScgiCcuZEYSCiWjMkwjIU19J28o2KmGAY6bhQV74vLfdANk5EDQJMYxrtL4AVB+X/BVLquFecAAABaxex0AwAAAIBe9/Nnf16BE2jue3PKTGXe3aYzyl7KKnc5p/zVvArXCspdzclMmPr5sz/f6WYDWGFKQYZSgGFx0KHW6wzDUBAEC9vSc1HKAQAAtBIzOQAAAIAGjdw5osf+82P6wv1f0Oxbs0oOJBVfG5dpmgqCYCEBuZN3lBxI6tOvflrrJtd1utkAVqBSAKJaoKHcddVmYEQtt5zpM9OaOjOfb2RvWuN7xpe9bstn6r61JOnC49XLXni8vnsuvl+5suXqrLeueuqMer/Far33cuVrKbu0XLvra2WdzXzn9bzvevu00d+Hdr2LZn+jUdvYjHs043cWQHUEOQAAAIAmGLlzRL9y9Vf0F//9X+hvvvg3KswUZMbDidO+4yvWH9O+X9ynD//mhzvcUgCoLGrwotZyJ4+c1LlT55RYm5AMLSzb5+Qd7Xhoh448d6TuNke15TONDXzXc03pXD2DnlEDPFHuV0v7ypWv1I9Rn6FaW5vdzqh1NvOd19pXzf4uGqmz2e+ikf7sRL9Uq7eef8cAiI4gBwAAANBED/zrB/TAv35A2emspl6d/wvlfWkNpgY73DIAqM3iAEVpGapmlLt4+qKOHTimgY0Dmtw/qeRgUoZpyHd82Vlb1oyl8y+e19N9T+voK0eV3peWVH1As5YBxEqDqc0ehKzW3k4PekZtX7kZBrU+V+lcvQPRS+/Z6nYuV+fS+9Zyfb11RtGsPq0map9GLddof0btl0p1Ry3bqcALsNqQkwMAAABogcHxQW1/cLu2P7idAAeAVe/i6Ys68eAJje8Z19YPbdXE/RNK700rtSulsXvHtOGuDRreOqzR7aMa2DSg4w8c19tff7ulbYoyoyLK4HiU+uqpsx7NaN/S62rpk2Y+e6vaWa2/6z1eS5311L/0vp0IlNXbp1HKNdKfneiXqN8NgOYiyAEAAAAAwApRWjKqWuLvcte1qtyxA8e0YdsGje8ZV3pvWps/sFnj941r7L1jGtk2ouEtw1o3sU6DqUENjQ8psSahZ/Y/U/Ge7dYNg8qdUutAbrv/an1pfd3azlp1y/teLGqf9vq7ANBbWK4KAAAAAIAVoJQMvBRwKLd8VKXrKiUUj1ru5JGTGhgb0Oj2UW28Z6NSe1Ia3jIsz/aUu5xTvD8uwzDku74825NruXLyjqw5S88//LwOfelQU/onqm7Kf4DWaMWsmSj1d6q+bgyudEo9fdPp7wbAuwhyAAAAAACwQpTLn7H0eKU8G80+d+7UOU3un9TApgENTQxpeMuw1k+ulzVjybM9OXlHdsZWcjAZbgNJJQYS6h/p12vPvdayIEc9icSbMYhZb1LpTi27g+YFAdr9jUXVDW2oRScDjrX0DcEjoHMIcgAAAAAAgJaYPjOtxNqEkoNJJdYmZCZMebYna8aSa7kK/PnZITFDZtyUmTBlJk3Fk/Fw64/r0tlLSu1ONdSOKEmjaz1fr6iJ0tulGwZmLzwevrPlEky3cqC73L0bDQK0+xurp65eDG61qr+i9k2rvhsAtSMnBwAAAADgFqVliWrJ0VDuunLnqt271rrxrrvvvlvT09MLW8niY0uPX758WS+//PLCsVbsv/TsS5IhGaahwA/k5BzlLueUmcoofzUvO2vLtVz5rq/AD6RgPr+HKRmxsMzUq1MN9Ex0zRz8rXWws1MDzt08GFsKdpS2diglsG4kkXUtfdru910tkXs3Bzxa/Y02o2+a8d0AiIYgBwAAAADgJktzLlQKRpS7rpQDorQtzddQbmmjSuWwvCeffFIHDhy4qd9KFh9benzxP1u1Hx6QfMeXk3VUuF5Q9lJWmamMcu/kVLhWUHGuKCfnyLVceY4XBjy8IAx6NMnSwcfFg5DLDV62YpmqWgMcnVqmqtsGZcsNFLd6ALnSYHczlxzrpn7vhjZU0sm+qrXOZnw3AKJjuSoAAAAAwC2WJqmu97pKuRtqqRf1SafTkY7fdtttC8cOHjzY/P1HDurNz74pO2urMFNQ35U+GYYhO2PLiIXJxp2co+JcUXbOlpO/OdhhmIbS+5Z/hmaotCySVHlwslUD2Y3WWY9uGmgvp5vbtpx6+7Sd77sX0QcAakGQAwAAAADQVWoNltw4f0Pf+Lff0JtffVPXz1+XJG3YtkFbf2Sr7v+v79fwHcMtbysqG98zLifvyJqxlL+aV7wvLt/1lRxMyoybCvxAruXKztkqzhZlZ+cDHXlXrhVuqV2N5ePolG4fnO329lXSrW3v1nb1KvoTQK0IcgAAAAAAWqq0BFWtFl9bruyLv/Ki/uaLfyMFUv9wv0buGJHv+cq/k9eZ3zujs8+c1d5f2KsP/toHm/IMiG7HQzt0/sXzYWAjFiYeTw4mZSZMKZA8x5OTd2Rnw0BHaVaHNWtp58M7O9LmRmdnRBmcbefSRvXer9YZL50ejI7azlK5RkTp02bdq1GVnr3RPo36zXTLN1VL3wDoPHJyAAAAAABapt4ARy3+8MN/qLNfPKvUrpS2fWSb7vzgnZq4f0Kp3Sml7ksp9f6UBjcN6mv/x9d04h+faGrdqN+R547IyTvKTGcW8nFk3s4oMxX+nHsnp/yVvArXCipcL6g4W5Q1a0m+dOjEoZa2rRUDlN0yOFtOo+1b2metGuQtd9+oSdzrzadRT53d/s6l6v0pta9Pq5XrliBPI32z+Fg3fxfASsFMDgAAAADALUrBiWqJvytdFyXAUa3M8QeO69K3LmnLwS0avWtUg+ODivfH5dmeirNF5a/llR/KK9YfkxE3dOGVCzrx0RPSvrqagSY7+spRHX/guDIXM3LyjhIDCcWTccmUAi+Q53hy8+7CDI4gCPTY6ceaVn+1QdVmz5CoVmenBj0bad/iv1pfrmy1AEC14/XmRCknajurlSunE+88ap9GDUi1qk+rvfN6+7Pd31rpXlG+GwDNRZADAAAAAHCTUtCiFLgot3xUtesW/3Px+cXHlpatlMD826e+rYv/+aIm90/qtvfdpk07N2nd5nWSIVkzlnKXc4r3x2WYhgI/kO/48mxP333pu9I6Sdsb7hpElN6X1qMvPKpn9j8ja85S/0i/4sm4jNj8u3J9uZa7MIPjsdOPKbWn9bk4+Avr+pRbnqcV/dhIXVHLtvP52q3S0kqrvU871TcAmocgBwAAAADgFuVmUyw9Xut1tZ6rdP6r/+tXNXLHiNZPrteG92zQ2PvGNLp9VIVrBWWmMgr8QJ7tySk4YY6HnB3meRgoyvlLhyBHh03cP6Enik/o1CdO6fUvv654f1yBPx/4Mg25lqudD++seYmqWgcfm63SPVs1qNms+zbjPvXeo5E6O1G2nc8X9Z7terZO1Ntr30uzygNoDEEOAAAAAEDXy05ndf38dW3Zv0VrRtaob7hPyYGkJMmMm4r1xRTvj9+6rYkrMZCQ3pKMXOWlt9Aeh589rMPPHtals5c09eqUpHCmR2pX62duAACAlYcgBwAAAACg602dmVoIZhgxQ77ry5qxJEMLSx35ri8F4RJYhmnIjJky4+FmxAwFU81NgI7GpHanlNpNYAMAADTG7HQDAAAAAADd4eDBg3rwwQd15syZha1k8bHvfOc7euqppxbOtWN/5h9mZMbMcEmqYphkPHspq8xURvnLeRVninJyjlzLled4CrwgXAppPugRM2LS9UZ6B+g+Wz5T+wYAwErFTA4AAAAAgCTpwIEDZc/dd999N/385JNPtnX/jT9/I5yxUXBVnCsqfzUvM2bKztoyY6Y825OdDXNwOPl3gx2+68t3fXm+J22o2gUAAADoMQQ5AAAAAABdL31fWr7nqzhXVOF6QYm1CQV+oGQmKTNuKvACOQVHdsYOt5wtN++GwQ7bU+AHCtIsV4WVhWTHAAAQ5AAAAAAA9IDB8UGNbhtV4WpBufU5mXFTnuMpOZhULBELl7GyPdk5W/bc/DY/q8MpONKopIFOPwUAAACajSAH/n/27j5GjvOw8/yvql9mOD1DDoek2NOUSFHWm80wlmhKceTlkeuXKMHGyYakEFkETgEV52LvHrQL3F6A7OkEBVofNn8E0F1ekEtOTBwuZYumFsHZZ2MJQ6ZX1joKGa1XOvqsgJaYDLtJcUjNsLurq6uqq+6Pnh4OR9Pd1dXvM98P0FCzqp56nn6q20ie3zzPAwAAAABD4dNf+bROPXFKiYmEDMOQV/YWQw5J8l1fru3KKTgq3yjLvmHLKTjySp70a31uPAAAALqCkAMAAAAAMBTu+5X7dOf+O3Xx+xcV+NXlqZKppGLJmGRIQaU6m8O1FoKO+bKsa5Y+8rmP6Cf3/ERitSoAAIBVx+x3AwAAAAAACOuJbz2h7f9ku+b/YV6FbEH5bF75bF6FXEGFKwUVrxRVvFqUNWupOFvURz7zET3+14/3u9kAAADoEmZyAAAAAACGypFvH9F3/+13dfaPz6pwpaDYSEzxker/e+vZnirlimLrYvrUv/mUDjx3oL+NBQAAQFcRcgAAAAAAhs5n/t1ntPe39uqNP35D7333PV37+2uSpKl7p3TXZ+7Sw//yYa2/fX2fWwkAAIBuI+QAAAAAAAylDTs26HP//nP9bgYAAAD6iD05AAAAAAAAAADAUCLkAAAAAAAAAAAAQ4mQAwAAAAAAAAAADCVCDgAAAAAAAAAAMJQIOQAAAAAAAAAAwFAi5AAAAAAAAAAAAEOJkAMAAAAAAAAAAAwlQg4AAAAAAAAAADCUCDkAAAAAAAAAAMBQIuQAAAAAAAAAAABDiZADAAAAAAAAAAAMpXi/GwAAAAAAQC8ZhrH4PgiCtq4zDKOlc2HrBgAAQDjM5AAAAAAArBm1kKEWMCwNHVq9rl7ZRtcHQbD4alQeAAAA4TCTAwAAAACwptSCi2ZBQ6PraoFFozBj+bkoMzdy53LKnstKkjJ7M5reM73idTteaPnWkqSLTzcve/Hp+ufqlW1Upp1yrWq3npXKhym7vFyv6+tWnZ1+bkvv1+p3Jup3rJ3y3XgWnf6OdvO316vfLQC0ipADAAAAAIAWtLpE1fLzNY2uO3n4pM6fOq/EWEIyJC1c6lqudj22S4dfPhyp7VHseGHlQcxGg8i1c50s16p266lXvl5/NKuzkWZt7XQ7o9TZ6ecWtq+i9mk7Ot2nnf4NNSvb7J6drq+Tv1sAiIKQAwAAAACAkNoJOKRbg42Vrp95fUbH9h9TanNK2/dtV3I8KcM05Lu+nIIje87WhdMX9PzI8zr62lFlHspIaj4wGWbwsdEgbKPB3V6Xa0Wzfgkz+Lz0fNj21c61OhBdL1joVjtXqnP5fZtd22p9UXWqT5uJ2qdRy7Xbp1H6pZO/2bBlAaCb2JMDAAAAAIAWGIax+Kr9O8y5ZmZen9GJz5/Q9J5p7fzsTm17eJsyezNKP5DWlo9u0dQ9U5rcOalN925SamtKxx89rktvXOrsh1um2eBqq4Oh7ZRrVZiB4VbvESYwiDqo3soAeLPrwi5Z1Mpz6ER/hq1/+b37MXDeap9GKddOn0bpl3Z+e518/gDQaYQcAAAAAIA1JWwAsdJ1SzcOX7pnR7NzYeo7tv+Ypu6e0vSeaWX2ZnT7J2/X9CemteVjW7Tx7o2a3DGp9dvWazw9ronpCSXWJfTivhdb/PRoJmwA0OtllJbXN6jtDGsQB8aj9umwPwsAGHYsVwUAAAAAWDNqG4LXAod6y0c1uq7dupf+u+bk4ZNKbUlp072btPn+zUrvSWtyx6QqTkXF94uKj8ZlGIZ8z1fFqcizPbmWK/uGrVcef0UHv3aw7fZhbej3/gn9DocGMVwBALSHkAMAAAAAsKbUCyyWHw8TbDS6ZqVz9a4/f+q8tu/brtTWlCa2TWhyx6Q2bN8ge85WxanItVw5eUfJ8WT1lUoqkUpodOOo3nr5ra6FHO1upN3NZYzawV/U39SJEKDVTcT7GTQMQhvC6FcY1M3nDwDdQsgBAAAAAEAf5c7llBhLKDmeVGIsITNhquJUZM/Z8mxPgb8wqyRmyIybMhOmzKSpeDJefY3GdfnNy0o/mG6rHY0GKsPu8dBKmXbKdcogDHRffLraDytt2tzNweN6925nsDvqBvfd0sv+7JZu9Vc3fnuD8HsCsDaxJwcAAAAAYE247777lMvlFl81S4+1cvxHP/rR4rEzZ85Efv+9l74nGZJhGgr8QG7RVfH9ovLZvKxZS07BkWd78j1fgR9IwcL+HqZkxKplsmez7XdQh7U7C6RbBvkv+WthR+3VC7XNqKNu8B22P3sdMDTbPH2QA49+fUej9Mkg/54ArB3M5AAAAAAArHrPPvusstlsS0tINTu+9Hw776sHJN/15RZcla6XFB+Ny7VcGYYhz/ZUvlGWW3Tl2Z4qbqUaeFSCaujRIfUGKevNMmhWtpvlohrUAdl6G1N3e8PqekFA2GfQasAxCP1e+3yDqld91Ynf3iA9VwBrGyEHAAAAAGBNyGQyHT2+9PyBAweiv3/igN79k3flFByV5koauToiwzDk5B0Zsepm427RVflGWU7RkWvdGnYYpqHMQyu3tRNaGfTuZ7lmhmFAdpDbtlyU/mwULgzD8+m2fvdBNwIuAOgFQg4AAAAAAPpoes+0XMuVPWfLmrUUH4nL93wlx5My46YCP5Bne3KKjsrzZTmFhaDD8uTZ1Vf6gfb241jthnlAdhDbPohtGnbD1KfD1FYAawN7cgAAAAAA0Ge7HttV3YPjqqXC5YLy2Xz1lcurcLmg4tWirFlLpQ9KKs+XF2d12PO2dj++u9/NH2itDsg2WyJqUAZ4o7az3XZH+fzL9/1YaQ+QqHuCtKrRbJJ2+zTqd2ZQvlNhDFNbAawdhBwAAAAAAPTZ4ZcPy7XcxVAjn80rf6kadBQuF1S8UpR11VLpWkml69Wgw563JV86eOJgV9vWbOC2V+WiaHdAdnlburWXQ7uBStR2rnRdozqHZYA7zHevV30a9vve74CnlfOD/vwBrD0sVwUAAAAAwAA4+tpRHX/0uPIzebmWq0QqoXgyLplSUAlUcSvyLG9xBkcQBHrq9ac6Vn+zAc5Gg969KteKpXWE+ev95cdqZVYq2+wv8psdj9onK7UjSjublVtJO/3Zjqh9GjWQ6lafNnvmrfZpN/ul020FgG5jJgcAAAAAAAMg81BGR75zRIXZgubem1tcsqqQKyzO5iheLSqfy8stuHry9JNK7+n+Xhz1lhFqNpjZ6XK91mgwu5d1hemvVo53os5B16j9/erTQTAsvz0AaBUzOQAAAAAAGBDbHt6mZ8rP6NQXTuntr7+t+GhcgR9IkgzTkGd72v347tBLVIUZsGxnUDNq2V4MpHaijlbv0Y++bKdsq/tqdEPU4KDd+3arfK/6tNf9QvgBYJARcgAAAAAAMGAOvXRIh146pMtvXlb2bFZSdaZH+oHuz9wAAAAYJoQcAAAAAAAMqPSDaaUfJNgAAACoh5ADAAAAAAAMvFY2kmZpHQAA1g42HgcAAAAAAAAAAEOJmRwAAAAAAGDgMTsDAACshJkcAAAAAAAAAABgKBFyAAAAAAAAAACAoUTIAQAAAAAAAAAAhhIhBwAAAAAAAAAAGEqEHAAAAAAAAAAAYCgRcgAAAAAAAAAAgKFEyAEAAAAAAAAAAIYSIQcAAAAAAAAAABhKhBwAAAAAAAAAAGAoEXIAAAAAAAAAAIChRMgBAAAAAAAAAACGEiEHAAAAAAAAAAAYSoQcAAAAAAAAAABgKBFyAAAAAAAAAACAoUTIAQAAAAAAAAAAhhIhBwAAAAAAAAAAGEqEHAAAAAAAAAAAYCgRcgAAAAAAAAAAgKEU73cDAAAAAKxdhmEsvg+CoOXrGpVfeq7VsgAAAACGAzM5AAAAAPRFLWSoBQwrhRLNrguCoG5AUTu30jWGYdxyrl7dAAAAAAYbMzkAAAAA9E0tfGgWNIS9rp5aqNGO3LmcsueykqTM3oym90yveN2OF6Ld/+LTzctefLr+uXbK1ivfrEwr6rUvbB1R27e8XK/r61ad7fZno/u1+l2J8t1qt3w3nkWnv6Pd7pd69+jk7xYAgGFAyAEAAAAADZw8fFLnT51XYiwhGZIWshLXcrXrsV06/PLhnrVlxwudH8BsNNDaqfqa1SFFG3xu1L6oYVM7/RGlnVHqbLc/W6k/ynWd1Ok+rVeunT4dpH5Zeo6wAwCwVhByAAAAAFjVVprFEWZGyMzrMzq2/5hSm1Pavm+7kuNJGaYh3/XlFBzZc7YunL6g50ee19HXjirzUEZS84HQMAOPjQZhmw3uRh3YrDeQ3qmgo1m/hBl8Xnq+1f5odSC61f5ot50r1bn8vs2ubbW+qDrVp81E7dOo5drt0373y/Lz3fwOAAAwSNiTAwAAAMCaVG+/DqkacJz4/AlN75nWzs/u1LaHtymzN6P0A2lt+egWTd0zpcmdk9p07yaltqZ0/NHjuvTGpa62txuDlY2Cl07W14lQZvl1YQKDqIPH7fRHq+1sFn612pZOfuaV7t2PQfNW+zRKuXb6dFD6pdlxAABWK0IOAAAAAH1Tm03RbFZF2OuitmF50HFs/zFN3T2l6T3TyuzN6PZP3q7pT0xry8e2aOPdGzW5Y1Lrt63XeHpcE9MTSqxL6MV9L3a8bYOiH8vxLK232WBur9u3vL5BbWdYgzgoHrVPh/1ZAACA1rFcFQAAAIC+qC0ZVQsulgYNS4OHZtctf1/vPkstLbf8/MnDJ5XaktKmezdp8/2bld6T1uSOSVWciorvFxUfjcswDPmer4pTkWd7ci1X9g1brzz+ig5+7WDkPumEqBtto/f6vXdCv8MhvptV9AsAAO1hJgcAAACAvqm3ZNRK/6533fJXo/s0u58knT8FGa0aAAAgAElEQVR1Xhvv3qjU1pQmtk1ocsekNmzfoLHNYxqdHNXI+hElx5M3X6mkEqmERjeO6q2X34rSDaG0s5H2IP/V+iC3rddqz6qdZ9bqJuL9HFAf9O9mTT/CoEZ1NpuNMgjPFgCAXmImBwAAAAAsyJ3LKTGWUHI8qcRYQmbCVMWpyJ6z5dmeAn9hVknMkBk3ZSZMmUlT8WS8+hqN6/Kbl5V+MN1WO8IMcIY512xw9uLTNwdUWy3baYMwINuv/ujGYHXUDe67pd/fr07oRn9F7Zel39Uw9wUAYDVjJgcAAACAnsvlcre8lh8/e/bs4rEzZ8707P33XvqeZEiGaSjwA7lFV8X3i8pn87JmLTkFR57tyfd8BX4gBQtLX5mSEauWyZ7NRumStjXboLrZ4GknZhG0apD/4rwf/VHbwDrqRtZh+3MQAqxWvpv91M3v6DD3CwAAg4SZHAAAAAB66rnnntM999yjAwcOfOjcSstHLT3W7ffVA5Lv+nILrkrXS4qPxuVargzDkGd7Kt8oyy268mxPFbdSDTwqQTX06JB6A6r1Zhk0u1eUpW+6vUHzoAYc/eqPegPeYZ95qwHHIPR7s+9mv/Wrr8L0S6O2RfnfCQAAhhkhBwAAAICee+edd/TEE0986Hgmk7nlv5JuCUO6/v6JA3r3T96VU3BUmitp5OqIDMOQk3dkxKqbjbtFV+UbZTlFR651a9hhmIYyD91se6e1Mugd5d69MkgD7fUMctuWi9KfjQbRh+H5dNsg90GztnXzfycAABhEhBwAAAAAsGB6z7Rcy5U9Z8uatRQficv3fCXHkzLjpgI/kGd7coqOyvNlOYWFoMPy5NnVV/qB9vbjGCTdGOgd5MHjZgax7YPYpmFHnwIAMFzYkwMAAAAAltj12K7qHhxXLRUuF5TP5quvXF6FywUVrxZlzVoqfVBSeb68OKvDnre1+/Hd/W7+hwzSckCtDh43WyJqUAajo7az3XZH+fzL9/1YaQ+QqHuCtCrKMmrLy9br06jfmUH4Tg3SbxYAgGFAyAEAAAAASxx++bBcy10MNfLZvPKXqkFH4XJBxStFWVctla6VVLpeDTrseVvypYMnDna1bc0GbhsdbzaoW+94pwZ7273f8nZ2e6+Qesdb3dQ7bDtXuq7Zvgth2tNv7Xw3690jap+2s89Fp3WjX5odBwBgtWK5KgAAAABY5uhrR3X80ePKz+TlWq4SqYTiybhkSkElUMWtyLO8xRkcQRDoqdef6lj9zQYpGw16d6O+di29f5i/3l9+rFZmpbJRw5tG5aP0R9R2Niu3knb6sx1R+zTq96tbfdrsmbfap4PWL/XqAwBgtWImBwAAAAAsk3kooyPfOaLCbEFz780tLllVyBUWZ3MUrxaVz+XlFlw9efpJpfd0fy+OessINRrMbLb0UKOB4kEZJG3Uxl7W1ay+qO0chmcQVTvfzUblu1WuVzrRL6v5ewMAQCuYyQEAAAAAK9j28DY9U35Gp75wSm9//W3FR+MK/ECSZJiGPNvT7sd3h16iKsygYzsDk/0q26v7t3qPYevLVvfV6IaowUG79+1W+V71ab/6pVP3AABg2BFyAAAAAEADh146pEMvHdLlNy8rezYrqTrTI/1A92duAAAAAGiMkAMAAAAAQkg/mFb6QYINAAAAYJAQcgAAAAAAWtLKhskspwMAAIBuYuNxAAAAAAAAAAAwlJjJAQAAAABoCbMzAAAAMCiYyQEAAAAAAAAAAIYSIQcAAAAAAAAAABhKLFcFAMACwzAW3wdBEOm6qOdaqR8AAAAAAABVzOQAAEA3A4ZauLA0cAh7nWEYCoJg8RX23ErnAQAAAAAA0BwzOQAAWFALF1YKIcJcFzWcqAUcYc3+f7P6x9f+Ue99/z1J0p3779Qdn7pDm+/fHKl+AAAAAACAYUXIAQDAAAgTlsxfnNeJz5/QBz/9QJIUH40rCAKd/8Z5GYahqbundORbRzRx+0RP2gwAAAAAANBvhBwAAHTB8tkZYWeHrFRWkn70lz/SN/+Hb2rD9g2645E7NDI+IplSpVyRU3Bkz9kq5Ap64SMv6Fdf/FXtPrK78x8KAAAAAABgwBByAADQYfWWn1oeZIT15v/1pv7T//SfdPvP366pu6c0nh5XYiwh3/PlFByVrpdkzVpKjCWUuJLQt778Lfme35HPAgAAAAAAMMgIOQAAWFALJ5oFEI2uC7O/Rit7cNhztr7529/UHY/coa0f36otH92iiW0TMuNmNeC4VlJyPKlYMla9rx8o8AL99VN/Lf2OpESoagAAAAAAAIYSIQcAALq5nFQtuKi3fFSz65b+d+n5RntuNNrA/OThk9pw+wZtvGujNt+3WekH0prcOSm35Kp4pahYIqbAD1RxK/JsT27JlWu5Gpkbkf2yLR1pr18AAAAAAAAGmdnvBgAAMCiCIFh8LT8e9rrlr2ZlGp33bE/vfu9drd+xXqnbUkptTWkiM6HUbSmNbhhVciKpxHhCiVSiulRV6ub7kQ0j0gXJ8MMviwUAAAAAADBsmMkBAMCAyp3LKT4aVzKVVHw0LsM05BQdWbOWKuVKdd+NQDJMQ2bMlBk3FUvEFEtWX0bcUJANtywWAAAAAADAMGImBwBgTbvtttv0qU99Sj/96U8XXzVLjy0/fu3aNZ08eXLxWDfef/vYt2WapmRIFbcip+Co+H5R+WxexdminBuOvJIn3/XlV6qBhwJJxkLwYZpSLnrfAAAAAAAADDpmcgAA1rQvfelL+uCDD1Y8t3HjxrrHR0dHtWvXrsVj3Xh/x913aLYyq0q5srjJeCwZk1t0ZZiGvLInJ+/IKTjybE+VckUVr6KgElQ3IA8CBSPM5ACGRaO9e8Je1+lzS483axcAAAAA9AMhBwBgzWsUZjQ6/rGPfWzxWDfe7/2VvXrz996UU3Bkf2Cr+H5RkuTkHRlxQ77ryyt5sudtOXlHruXeDDvcSnUwMtP4swMYDLUwIQgCGYYhwzBWDBQaXdfs3PLwIky52nEAAAAAGFSEHAAADKjN929WoED2dVvWrKXYSEy+5ys5npQZNxX4gbyyJ7foyp63Vc6X5RZduSVXXslTYAQyNrPxODAsamFCLWyIcl3Ye0Stu/FNpPfOvKd3v/uuZn44I0m6/ZHbtfOf7tSd+++U+J8jAAAAAF1AyAEAwIAyTEOf+M1P6M2/eFOJ8YQM01ClXKmGHAlTCqp7dXglrzrbY95W+UZZTsFROV+WPiEF4i+wAXw4vGhldkaYcv/4g3/UN77wDdlztgzTUHIsqcAPNPPDGf3wD36o0alR/frLv67MzzG9DAAAAEBnEXIAADDAfvF//0W9/bW3lb+UlyR5tqdkKikzacowDPmeX53NYbly8tWgo/RBSaMbRlV4tCAyDgBS4+WqGllp747lx37w+z/Q93/v+5q8a1Jbd29Vcjwpw7i5b1BprqTilaKOffqYPvuVz+rnnv65znwoAAAAABAhBwAAA+/L/++X9Se7/0TzF+flWq4SqYRiyZgM01DgB6o4C7M5itW9OxJjCX357S/r9//w9/vddACr3JnfO6Mf/sEPdfsjt2vq7imNp8eVWJeQ7/kq58sqXS9pZHZEiXUJxdfF9er/8qpcy+13swdCPzeaBwAAAFYTQg4AAAbc2JYxffnHX9af7vlT5S/lFRuNKTmWlBGr/kW17/pyio5819f41nF98ewXNTo52u9mA2hRbZZEsz0xGl0X9h6dKDf37pxe+99e0x2P3KGtP7tVWz62Reu3rZcRM+TkHVnXLCVTScUSMUlS4AfyPV/fe+570r+QNNFSE1eVTm0032gz+SgzdwAAAIBhRMgBAMAQWLdxnf7Vu/9K//kr/1lv/OEbmv/HeSVSCUmSW3Q1nh7XJ3/nk3rkdx7pc0sBRLF0ELv275qlA9SNrgtzbum/w9a9vJ01//G//4/acMcGTe6c1KZ7Nyn98bQ23rVRTsFR4UpBRtxQUFmYbWZ78kqe3KIrZ86R9YolPRmxs1aJdjea73RokTuXU/ZcVpKU2ZvR9J7pFa/b8UK0+198unnZi083v89K92hUrl6dYepqRbv1tPq56pXrdX3dqrPTz23p/Zrdo9U+bfd73atn0envaNR+6fRvDwAAiZADAIChsu9392nf7+5TpVy5ZTAqloz1uWXohn4vZ7PS+UaD3mhPvb5cfrxRn3f6XL3jTsHRzA9ntH3fdo1tHtPYljGlbktpdOOoZEiJQkLJsaQSY4mbr3UJxcfiSqxPSP8gGV5rs03wYc2Cq5XOLXfy8EmdP3VeibGEZGhxLyfXcrXrsV06/PLhjre7nh0vRBukrVeu0eBz7VwnBlzbrafVz9WszkaatbXT7YxSZ6efW9i+itqn7eh0n3bjtxClX3r12wMAoIaQAwCAIRQbiemOR+7odzPQRZ1azqbRuUbL2TRa3oZgA7lzOcVGYtXgYiQuQ4bKhbKKV4qqOBX5rl/9nhiSYRoyYobMhKlYIqZYIiYjZsi/5Pf7Ywy9dparmnl9Rsf2H1Nqc0rb922vbhhvGtUlEAuO7DlbF05f0PMjz+voa0eVeSgjqflAaJiBy0aDsGEGaZeeb1Yuan2tatYv3fhcS8u0OhBdL1joVjtXqnP5fZtd22p9UXWqT5uJ2qdRy7Xbp632Sz+fIQBg7TH73QAAAACsbOkyNVGvC3uPpTq5fr9bdHXxzEX94Pd/oB/8/g908fsX5RbZeLrmzjvv1Kc//Wm98847euedd5TNZvWXf/mXi+cH9f2V/3ZFpmlKhlRxKyrnqwFHPptXcbao8nxZruVWA4+Kr8APbgk9TNOUcYWZHP0y8/qMTnz+hKb3TGvnZ3dq28PblNmbUfqBtLZ8dIum7plaXIYstTWl448e16U3LnW1TWEHO5df12xQtpXgI6owA8Ot3iNMYBB1UL2VAfBm14VdsqiV59CJ/gxb//J792PQvdU+jVKunT6N0i+dfIYAAITBTA4AAAB8SDvLXNX8h1/8D/rpd3+qWDImwzSkoLr5dMWt6K7P3aUj/8+Rzjd8yDz55JPK5/OL/47H49q/f//ivwf1/djmMfkVX57tyck7Kl0rKZaMyS26MkxDXrl63Ck48krezdkdXqCgUg08grG1PSOo3Y3m2wkjj+0/puk905reM63N92/WxLYJmQlTbtFV6XpJI1dHFB+Jy4xV/yYuP5PXi/te1DPlZyLV164wA+U7Xhi+vwwf1M+1vL5BbWdYg9imqH067M8CAIBuIeQAAABYg8Judix9eOPrpVYaaH339Lv66i98VRvu2KA7PnWHkuNJmaapiltZXAbn0t9c0nPmc/qNV39DO/bv6OAnGz4TExO3/PvOO+8c+PeZvRkFQVANOK6XlBxPVv894VSXovJ8eSVP5RtllfNluUW3uvl42VPFrcj3fRnb1u5Mjk5uNL/032HOnTx8UqktKW26d5M2379Z6T1pTe6YVMWpqPh+UfHRuAyj+gxrm8a7liv7hq1XHn9FB792sMO9gdWq33sv9HqPjaibwAMAgPYRcgAAAKxRywdWO+Gnp3+qbzz+Dd3xqTs09ZEpjafHlRhLyK/cXOffmrWUSCVUvFLU1/751/Trr/x6R+pG70zunFRyPCn7evV5xpIx+Z6vcr6sWDymwA/klT25RXcx6HCKjtySK7fkSiOSJvv9KfqrXxvNnz91Xtv3bVdqa0oT2yY0uWNSG7ZvkD1nq+JU5FqunLyj5Hiy+kollUglNLpxVG+9/FbXQo5+DUh3eyC6H5tZD6pOhACtbiLez6BhENoQRr/DIAAAOoGQAwAAYEC1u5xN2Ht0cg+Ov3r0r3THJ+9Q+uNpbf7oZk1kJhRLxOQUq0saFa8Wb12+ygv01c99VerPKjiIyIybeuhLD+m//MF/UWI8IcM0VHEqSqQSiiVjUlDdq8MreXIKjsrzZZVvlOUUHLkFV/p5KTDW9nJV/ZA7l1NiLKHkeFKJsYTMhKmKU5E9Z8uzPQX+wsyRmCEzbspMmDKTpuLJePU1GtflNy8r/WC6rXY0GuTs1oDwSnX2cvB5EAa6Gy1l1M2B53r3bicEiLrBfbf0sj+7pdff0UH4TQAAVg9CDgAAgAHUyeVs6t1j6XUr1b3S+eVhydJzX/vVr2kiM6GNd2+sLoPzYFqTd06qYi8sgzNS/T89b1kGp+QqMZ+Q87IjPdZKD6HfDjx3QD/66o90Y+aGFEie7S2GHLXljryytzgzoDxfVul6SROZCV3/765LazTjyOVyt/x7enq65eM/+clPdN9990mSzpw5s7hXSrP32XPZxc3fAz+QW3RVfL+oilNR4AfVPVRsT75X3SxewcJv3pSMWLVM9my27ZBjUHR734JB/kv+fgzCtxsEhO3PXn+2epun19oxyPtj9Po7Osi/CQDAcDP73QAAAACsLAiCxdfy42Gua3aPemXClrslUPED/eT//omm7prS+NZxjU+Pa8P2DVq/bb3WbVqnkQ0jSk4kq6/Ura91k+uk8y11DQbEl97+kmLJmObem1M+m7/5yuVVuFJQ8f2irKuWStdKKrxf0Mj6Ef32f/vtfje7b5577jm9+uqrK/+G6v22Vji+/Hwr7xVIvuvLLVQ3GS9cLiifzat4pajStZLKN27uoVJxK9XAoxJUQ48Oufj0yi+pewPUva5vUAdzl372MMc7WW+9Y2GeQasBxyD0+yC0oRECDgDAasJMDgAAALQtdy6nxLqEEuOJ6jI4cVNeyVPpg5IqduXmMjjmzWVwYolY9ZWMSQnJyK3djaiHVTKV1L/+h3+tP/u5P9P1v78uwzSqszniMQVBIN/x5ViOFEi37bpNv/nD35TW+GN+55139MQTT3zoeCaTWfH6lY7ff//9i+8PHDgQ+n1mb/VeTsFRaa6kkasjMgxDTv7mhvG1fVScoiPXujXsMExDmYdWbmcnNFpOaZjqG4bB3EFu23JR+rNRcDIMz6fbCDgAAKsNMzkAAAAGiOu6unjxomZmZhZfP/7xjxfPnz59eiDf5/4uJ8Oshhh+pTpQal21lL+UlzVryckvLIPj3lwGR4YWl8GJGTEp27x/MHiMmKHfOvtb+qUXfknrb18v66ql4mxR1jVL1nVLG7Zv0C/94S/pN/+GgKPfpvdMy7Vc2XPVDeNrszjy2bwKuYKKV4qyZi2VPiipPL+wh4rlyrM8eXb1lX5gdSxV1S3DPJg7iG0fxDYNOwIOAMBqxEwOAACAAfKVr3xFd911l37hF35h8djIyMji+1QqNZDvzbipwA9UcSrVvxK/XlJ8NC6n6MgwDXm2p3K+fPMvw51bl8EJFPDnN0Pu47/xcX38Nz4uSbr+99clSVP3TPWzSVjBrsd26cLpC0qOJ2XGqhuPJ8eTMhPm4obxruXeumF80ZE9b2v347v70uZmMy4GZRC11XYMy+eK2s6l+1JEEeXzN7q2X4P7K2m3T6N+Zwg4AACrFf+vJAAAwIC5cOGCtm7duvi66667Fs898sgjA/l++hPTUlBdBsf+wFbxavGWvxC3rlqyr9uy5+3qX4eXloQdrl/dbyCzRneiXoWm7pki4BhQh18+LNdyq/um1GZyXFr4rV5emM2xsI9K6Xp1Roc9b0u+dPDEwa62rZW9GcKUaXa/Tu7H0e5gbiufqx317ht1U++w7VzpukZ1DsvgeLP+lHrXp2G/7wQcAIDViJkcAAAAaFv6gbRc21V5rixr1lJsJCbf85XMJxdneXhlT27BrQYdeae65v9C2BF4gbS1358CWBuOvnZUxx89rvxMXq7lKpFKKJ6MS6YUVAJV3Io8y1ucwREEgZ56/amO1d9sMLbeJtW1ciuVb/aX663W14qldTT76/2VjnXycy0/3ihAaEXUdjYrt5J2+rMdUfs0aiDVrT5t9sxb7dMo/dKvZwgAWLuYyQEAAICO+NknflY3sjdUvFr80Fr/hcsFWe9bsmYt2R/Yi0GHW3BVzpelj/e79cDakXkooyPfOaLCbEFz78196LdavFKszsbK5eUWXD15+kml93R/L46LTzce9Gw06NvK8bD19UrU9ne6rjD91crxTtQ56Jp9X/vRpwAArEXM5AAAAEBH/NrxX9NXxr6iQrYgBZJne0qmkoolY5Ih+Z4vz/aq6/3nHdlz1bDDMA3pn6u6GTmAntj28DY9U35Gp75wSm9//W3FR+MK/OqPsLaPzu7Hd4deoirM4GonBmBbvUcvBn2H7XP1o2yn9tVoR9TgoN37dqt8r/o0SlnCFgBArxFyAAAAoGO++Ldf1F/s/wvdmLkht+QuhhyGaSwug+OWbm5qbBiGvvi3X9Qfff2P+t10YE069NIhHXrpkC6/eVnZs1lJ1Zke6Qe6P3MDAAAA6ARCDgAAAHTMll1b9OT3ntSf//yfq5wva92GdYqNVkMOBVLFq6hSqqhcKMuMm/riG1/Upvs29bvZwJqXfjCt9IMEGwAAABg+hBwAAADoqNt+5jb9bv539c0vflP/9a/+qwzDkGEYChRIgRQEgR588kH9sz/9Z/1uKgB8SCsbSbMsDwAAQP8RcgAAAKArfvnPflm//Ge/rOt/f/2WZXCm7p7qc8sAAAAAAKsFIQcAAAC6auqeKU3dQ7ABYDgwOwMAAGC4mP1uAAAAAAAAAAAAQBSEHAAAAAAAAAAAYCgRcgAAAAAAAAAAgKFEyAEAAAAAAAAAAIYSIQcAAAAAAAAAABhKhBwAAAAAAAAAAGAoEXIAAAAAAAAAAIChFO93AwAAAACsXYZhLL4PgiDSdc3uUe98mLoNw2jYLgAAAAD9xUwOAAAAAH1RCxlqIcLS0CHsdc3uUQspaq96x1equ157AAAAAAwOQg4AAAAAfVMLHprNlmh0Xb1zjWZhNKuPGRwAAADAcGC5KgAAAACrVpRlrloJOD648IH+9o/+Vu+++q6uX7guSZq6e0o7/+lOPfw/PqzJOyfbaT4AAACAJgg5AAAAAKxajcKLlc61EnCc/p9P6+/+/O+kQBqdHNXGOzfKr/iyrlg693+e05svvqm9X9qrz3zlM537QAAAAABuQcgBAAAAAEvU29djqb/63F8pdy6n9ANpjW8dVzKVlCS5tqvyjbLs67asWUt/83/8ja68eUVPfPuJnrUfAAAAWEsIOQAAAAD0zdIZFFGvC3uPlcosF2Zfj+OPHtflH13WjgM7tOmeTRqfHld8NK6KU1F5vizrmiVrwlJsNCYjbujiaxd14pdPSA+Fbh4AAACAkAg5AAAAAPRFLZiohRP1lpZqdF2Yc0v/HeZcIz8+9WPN/HBG2/dt120/c5u27t6q9bevlwzJnrNVfL+o+Ghchmko8AP5rq+KU9F733tPWi/p3pCdAwAAACAUQg4AAAAAfVMvXFh+vFEI0Y1z9a559X99VRvv3KgN2zdo6iNT2vIzW7Tp3k0qXSspn80r8ANVnIrckivXcuUUHTkFR+VUWe53XUIOAAAAoMPMfjcAAAAAAIZBIVfQ9QvXNXbbmNZtXKeRyZHFvTjMuKnYSEzx0fiHX+viSqQS0jXJKIZfUgsAAABAc8zkAAAAAIAQsueyi2GGETPke77sOVsyJN/z5dmefM+XgupyW4ZpyIyZMuPVlxEzFGTDLYsFAAAAIBxmcgAAAADoqQMHDujzn/+8zp07p7feekvPPffc4rlBfv/SH70kM2ZWl6QqVzcZL1wuKJ/Ny3rfUnmuLLfoyrM9VdyKgkqgwA8WQ4+YEZOuN+ud4VfbIyXMZvIrXdeo/NJzjcqFqR8AAACrAzM5AAAAAPTU/v37b/n37t27F98/++yzA/v+C//iC/rG978hr+SpfKMsa9aSGTPlFByZMVMVpyKnUN2Dw7Vuhh2+58v3fFX8ijS1YpesGks3gK8FDSvtfdLoutp/64UUYfdxIeQAAABYGwg5AAAAACCEzCcy8iu+yjfKKl0vKTGWUOAHSuaTMuOmgkogt+TKyTvVV9GRZ3nVsMOpKPADBZnVv1zV0rCiUdAQ9roo6oUry+XO5ZQ9l5UkZfZmNL1nesXrdrwQrR0Xn25e9uLT4e610n3qla1XZ9i6wmq3nlY+U6Nyva6vW3V2+rktvV+ze7Tap+1+r3v1LDr9HW3lWfT6+w0AaxkhBwAAAACEMD49rk13b1JptqTihqLMuKmKW1FyPKlYIlZdxsqpyCk6cm4svBZmdbglV9okKdXvTzH8lgYiYYKMlZw8fFLnT51XYiwhGZIWbuNarnY9tkuHXz7cgZaGs+OFaIO09co2u17qzKBpu/XUK9+oP6KGTa32YZiyg/7cwvZV1D5tR6f7tF65dvq0nX6J+n3rx7MAgNWCkAMAAAAAQvr0Vz6tU0+cUmIiIcMw5JW9xZBDknzXl2u7cgqOyjfKsm/YcgqOvJIn/VqfG78KrLQkVZhjNTOvz+jY/mNKbU5p+77tSo4nZZiGfNeXU3Bkz9m6cPqCnh95XkdfO6rMQxlJzQclwww+NxqEbTa426x8mGtbrS+MqPXUm2EQtn21c60OCtcLFrrVzpXqXH7fZte2Wl9UnerTZqL2adRy7fZp1H6pV3eY+7RTJwCsVWw8DgAAAAAh3fcr9+nO/Xdq/uK88rm88tm88pcW/pvLq3ClIOuqpdK1kuwPbJXnyrKuWbrrs3dJ9/S79WvbzOszOvH5E5reM62dn92pbQ9vU2ZvRukH0try0S2aumdKkzsnteneTUptTen4o8d16Y1LXW1T2CV96l3XyqBumPpa0Yl6ll8XJjCIOqjeal81um7Qn1ur4Vs/lkNqtU+jlGunT6P2S5Rn326dAABCDgAAAABoyRPfekLb/8l2zf/DvArZQjXgyOZVyBVUuFJQ8UpRxatFWbOWirNFfeQzH9Hjf/14v5vdM7XlpJrtsxH2uk45tv+Ypu6e0vSeaWX2ZnT7J2/X9CemteVjW7Tx7o2a3DGp9dvWazw9ronpCSXWJfTivqrvgwMAACAASURBVBd70ra1JOwgcK//in15fYPazrAGcbA8ap8O+7MAAHQfy1UBAAAAQIuOfPuIvvtvv6uzf3xWhSsFxUZiio9U/98rz/ZUKVcUWxfTp/7Np3TguQP9bWwP1TYRrwUXS5eNWrqMVLPrlr+vnV8eiIRdqurk4ZNKbUlp072btPn+zUrvSWtyx6QqTkXF94uKj8ZlGIZ8z1fFqcizPbmWK/uGrVcef0UHv3Ywcp9E1cn9M9A7/X5u/Q6H1vL3td/PHgDWMkIOAAAAAIjgM//uM9r7W3v1xh+/ofe++56u/f01SdLUvVO66zN36eF/+bDW376+z63svXr7YSw/Hva6sOcanT9/6ry279uu1NaUJrZNaHLHpDZs3yB7zlbFqci1XDl5R8nxZPWVSiqRSmh046jeevmtroUcrQxId2IwuVcD4PxF/U29fG6DMMg+CG0IoxffUQIgAOgdQg4AAAAAiGjDjg363L//XL+bgQZy53JKjCWUHE8qMZaQmTBVcSqy52x5tqfAX5hVEjNkxk2ZCVNm0lQ8Ga++RuO6/OZlpR9Mt9WORoOqjQY/65VrZzC5V4OtgzCoe/Hpal+ttMF0Nwe6+/XcetnnvezPbulGf3Xj2QMAGmNPDgAAAABA23K53C2vpcfPnj27+O8zZ8709H32XFYyJMM0FPiB3KKr4vtF5bN5WbOWnIIjz/bke74CP5CChWWxTMmIVctkz2Yj9Ukn1TYl7taGyJ0yyAO5tbCj9uqFXj23XgcMzTZPH+TAo1ff0XafPQAgPEIOAAAAAEBbnnvuOb366qsKgmDxVdNomaqevQ8k3/XlFlyVrpdUuFzdML54pajStZLKN8pyi251PxW3Ug08KkE19OiQ5QOeSwc+mw0INxpQDjOYvNYDjnqDzN0efO7Vcxukfh+ENjTSy4Cj3rFBDoAAYFixXBUAAAAAoG3vvPOOnnjiiQ8dz2QyymQyi/8+cOBAT9/n1ldnlTgFR6W5kkaujsgwDDl5R0asutm4W3RVvlGWU3TkWreGHYZpKPPQzfZ3WqPllDphrQccSw1y25aL0p+NBs+H4fl0G30AAKsXIQcAAAAAYNWa3jMt13Jlz9myZi3FR+LyPV/J8aTMuKnAD+TZnpyio/J8WU5hIeiwPHl29ZV+oL39OPqFgKO5QWz7ILZp2NGnALC6sVwVAAAAAGBV2/XYruoeHFetxaWq8tm88rm8CpcLKl4typq1VPqgpPJ8eXFWhz1va/fju/vS5nYHYwc14Gi2ZM+gDEZHbWc/nlu9pdCW3qNX+0I0mk3Sbp9G/c70c4kqAEBvEHIAAAAAAFa1wy8flmu5i6FGPptX/lI16ChcLqh4pSjrqqXStZJK16tBhz1vS7508MTBrratlb0ZVjq20sDqoAYc9crX+3entBuoRG3noD63djXrT6l3fdqsXL/6tNVnDwBoD8tVAQAAAABWvaOvHdXxR48rP5OXa7lKpBKKJ+OSKQWVQBW3Is/yFmdwBEGgp15/qmP1R91cvFYuyiBwmL+qj6qdepp9rmZ/kd/seKMAoRVR2znIz61RvY2OL683aiDVrT5t9sxb7dOo/RLl2bdbJwCAmRwAAAAAgDUg81BGR75zRIXZgubem1tcsqqQKyzO5iheLSqfy8stuHry9JNK7+n+XhzNlhFqNOg7zIOdjT5XL+tqVl/Udq7W5yY1/772o08HyWp+9gAwqJjJAQAAAABYE7Y9vE3PlJ/RqS+c0ttff1vx0bgCP5AkGaYhz/a0+/HdoZeoCjNg2YlBzVb3Z+iFXn+uduvsR9lBeG5Rg4N279ut8r3q0358PgIQAIiOkAMAAAAAsKYceumQDr10SJffvKzs2ayk6kyP9APdn7kBAACAziLkAAAAAACsSekH00o/SLABAAAwzAg5AAAAAKDHDMNYfB8EQaTrunEOa1crmySzrA4AABgkbDwOAAAAAD1UCxlqAcPS0CHsdYZhKAiCxVcnzgEAAADDiJkcAAAAANBjteCiWdBQ77pGMzA6PTsjdy6n7LmFfSv2ZjS9Z7qj98dgYHYGAAAYVoQcAAAAAIAPOXn4pM6fOq/EWEIyJC1kJ67latdju3T45cN9bR8AAAAgEXIAAAAAwFCrLUEV5lyYJapmXp/Rsf3HlNqc0vZ925UcT8owDfmuL6fgyJ6zdeH0BT0/8ryOvnZUmYcyHf08AAAAQCsIOQAAAABgSLUScNQsPbY88Jh5fUYnPn9C03umteneTUptTSkxllDgB3ILrkpzJVmzlpLjSeVzeR1/9LiOfOdIZz8UAAAA0AJCDgAAAADosVoA0WxWRaProgQcza45tv+YpvdMa3rPtDbfv1kT2yZkJky5RVel6yWNXB1RfCQuM2ZKkvIzeb2470XpdxtWBQAAAHQNIQcAAAAA9FAttKgFF8tnVizfbLzedUv/u/R8mHPL7ydV9+BIbUlp072btPn+zUrvSWtyx6QqTkXF94uKj8ZlGIZ8z1fFqcizPbmWK/uGLeekI7FFBwAAAPqAkAMAAAAAeqzeLIvlx8Ne14lz50+d1/Z925XamtLEtglN7pjUhu0bZM/ZqjgVuZYrJ+8oOZ6svlJJJVIJjW4clXPeqXtfAAAAoJvMfjcAAAAAANBfuXM5JcYSSo4nlRhLyEyYqjgV2XO2PNtT4C/MKokZMuOmzIQpM2kqnowrnoxLccm43HjpLQAAAKAbCDkAAAAAoEfuu+8+5XK5xVfN0mO5XE7vvffe4rkzZ850/X32XFYyJMM0qpuMF10V3y8qn83LmrXkFBx5tiff8xX4gRQsLH1lqhp8GKaUbaNjAAAAgIhYrgoAAAAAeuDZZ59VNptdccmoRstU9ex9IPmuL7dQ3WQ8PhqXa7kyDEOe7al8oyy36MqzPVXcSjXwqATV0AMAAADoE0IOAAAAAOiRTCbT0nFJOnDgQNff59ZXZ5U4BUeluZJGro7IMAw5eUdGrLrZuFt0Vb5RllN05FrLwo4gUJAh7AAAAEDvEXIAAAAAwBo3vWdaruXKnrNlzVqKj8Tle76S40mZcVOBH8izPTlFR+X5spzCQtBhedU9O7xASvf7UwAAAGAtIuQAAAAAAGjXY7t04fSFarARq248nhxPykyYUiBV3Ipcy5VTqAYdtVkd9rwt7ep36wEAALBWEXIAAAAAAHT45cN6fuR55XN5SZJne0qmkjKTpgyjumSVZ3tyreqyVeX5cjXg8CUdlsRqVQAAAOgDs98NAAAAAAAMhqOvHZVbcJWfySufzetG9oYK2YLyubwKlwsqXi3KumqpdL0k67qlIAj01OtP9bvZAAAAWMMIOQAAAAAAkqTMQxkd+c4RFWYLmntvTvlsNewo5ArVkONKUcWrReVzebkFV0+eflLpPWzGAQAAgP5huSoAAAAAwKJtD2/TM+VndOoLp/T2199WfDSuwK+uRWWYhjzb0+7Hd+vgiYN9bikAAABAyAEAAAAAWMGhlw7p0EuHdPnNy8qezUqqzvRIP8DMDQAAAAwOQg4AAAAAQF3pB9NKP0iwAQAAgMFEyAEAAAAA6BjDMBbfB0HQ8nXNyocp16xuAAAArB5sPA4AAAAA6Iha0FALGJYHD2GuC4KgbkBhGMbi+SAIVgw2GpUHAADA6sNMDgAAAABAx9QChpVCiCjXdVPuXE7Zcwv7jezNaHrP9IrX7Xgh2v0vPt287MWnw91rpfvUK1uvzrB1hdVuPa18pkblel1ft+rs9HNber9m92i1T9v9XvfqWXT6O9rKs+h1WwFgLSPkAAAAAACsCmGXyjp5+KTOnzqvxFhCMiQtXOparnY9tkuHXz7c5ZbetOOFaAOf9co2u17qzKBpu/XUK9+oP6KGTa32YZiyg/7cwvZV1D5tR6f7tF65dvq0nX6J8n3r1e8WAFYrQg4AAAAAwFBoNOtjeahRW9pqqZnXZ3Rs/zGlNqe0fd92JceTMkxDvuvLKTiy52xdOH1Bz488r6OvHVXmoYyk5gOhYQYfGw1sNhvcbVa+G/WF0axfwgzoLj0ftn21c60ORNcLFrrVzpXqXH7fZte2Wl9UnerTZqL2adRy7fZp1H6pV3erv9mlZbr5/AFg2LEnBwAAAABgaETdd2Pm9Rmd+PwJTe+Z1s7P7tS2h7cpszej9ANpbfnoFk3dM6XJnZPadO8mpbamdPzR47r0xqUufYqqsEv61Luu3qBuK9dHFWZguNV7hAkMog6qtzIA3uy6bj23sPU302r41o+B81b7NEq5dvo0ar9EefbN2kOwAQDNEXIAAAAAADqmNtOi2T4bYa9rVL6VoOPY/mOauntK03umldmb0e2fvF3Tn5jWlo9t0ca7N2pyx6TWb1uv8fS4JqYnlFiX0Iv7XozUNtQXdhC418soLa9vUNsZ1iAOjEft02F/FgCA7mO5KgAAAABAR9SWk6oFF0tDiKWhRLPrlr+vnW+058bysGTp+ZOHTyq1JaVN927S5vs3K70nrckdk6o4FRXfLyo+GpdhGPI9XxWnIs/25Fqu7Bu2Xnn8FR382sHonRIR6/APp34/t36HQ2v5+9rvZw8AaxkhBwAAAACgY+rNrlh+POx1nTh3/tR5bd+3XamtKU1sm9Dkjklt2L5B9pytilORa7ly8o6S48nqK5VUIpXQ6MZRvfXyW10LOVoZkO7EYHKvBmH5i/qbOvncwl7Xz0H2QWhDGL34jnYqAOL3BADNEXIAAAAAAFat3LmcEmMJJceTSowlZCZMVZyK7Dlbnu0p8BdmlcQMmXFTZsKUmTQVT8arr9G4Lr95WekH0221I8qGw43KhRlMXqlsLwefB2Gg++LT1X5YadPmbg4et/Pc6om6wX239LI/u6Ub/dWNZ99OOQBYC9iTAwAAAADQtvvuu0+5XG7xVVP795UrV3TmzJnF4716nz2XlQzJMA0FfiC36Kr4flH5bF7WrCWn4MizPfmer8APpGBh6StTMmLVMtmz2cj90im1jZDb3Si62wPRg/yX/LWwo/bqhXafW9j+7HXA0Gzz9EEOPHr1He3Eb3aQf08AMEgIOQAAAAAAbXn22We1f/9+BUGw+KpZemz58V69VyD5ri+34Kp0vaTC5YLy2byKV4oqXSupfKMst+jKsz1V3Eo18KgE1dCjQ5YPeC4d+Gw2INxoQLnZDJEo9UU1qAOy9QaZ2w2MwtRb71iYZ9BqwDEI/T4IbWiklwFHvWPDtPwYAAwLlqsCAAAAALQtk8k0PZ5O31zy6cCBAz15n9lbrd8pOCrNlTRydUSGYcjJOzJi1c3G3aKr8o2ynKIj17o17DBMQ5mHVv5sndBoOaVhqm8YBmQHuW3LRenPRoPnw/B8um2Y+mCY2goAg4CQAwAAAACwak3vmZZrubLnbFmzluIjcfmer+R4UmbcVOAH8mxPTtFReb4sp7AQdFiePLv6Sj/Q3n4cq90wD8gOYtsHsU3Dbpj6dJjaCgCDguWqAAAAAACr2q7HdlX34LhqLS5Vlc/mlc/lVbhcUPFqUdaspdIHJZXny4uzOux5W7sf392XNg/LAGerA7LNluwZlAHeqO1st91RPn+9pdCW3qPbS3PVNFs+rdE1zfo06nemn0tUtWpQvv8AMGwIOQAAAAAAq9rhlw/LtdzFUCOfzSt/qRp0FC4XVLxSlHXVUulaSaXr1aDDnrclXzp44mBX29bK3gwrHVs+GNrsfp3cj6PdAdnlben2XiH1jre6qXereyqErXNYBrib9afUuz4N+33vdZ+2+uzDnAcA1MdyVQAAAACAVe/oa0d1/NHjys/k5VquEqmE4sm4ZEpBJVDFrcizvMUZHEEQ6KnXn+pY/VE3F6+VayUACHNtp2YcNKsvyudqNgjc7HijAKEVUdsZ5bm105/tiNqnUQOpbvVps2feap9G7ZdO/GZ7+fwBYLVgJgcAAAAAYNXLPJTRke8cUWG2oLn35haXrCrkCouzOYpXi8rn8nILrp48/aTSe7q/F0ezZYQaDfrWCxDaqa9XGn2uXtYVpr9aOd6JOgdds+9rP/p0kKzmZw8Ag4qZHAAAAACANWHbw9v0TPkZnfrCKb399bcVH40r8ANJkmEa8mxPux/fHXqJqjADlp0Y1Gz1Hr0YSB22z9WPsq3uq9ENUYODdu/brfK96tNefz7CDwBoDyEHAAAAAGBNOfTSIR166ZAuv3lZ2bNZSdWZHukHuj9zAwAAAJ1FyAEAAAAAWJPSD6aVfpBgAwAAYJgRcgAAAAAAsMa1skkyS+sAAIBBwsbjAAAAAAAAAABgKDGTAwAAAACANY7ZGQAAYFgxkwMAAAAAAAAAAAwlQg4AAAAAAAAAADCUCDkAAAAAAAAAAMBQIuQAAAAAAAAAAABDiZADAAAAAAAAAAAMJUIOAAAAAAAAAAAwlAg5AAAAAAAAAADAUCLkAAAAAAAAAAAAQ4mQAwAAAAAAAAAADCVCDgAAAAAAAAAAMJQIOQAAAAAAAAAAwFAi5AAAAPj/2bv/GDnO/M7vn6rp7hmxh+SQQ5rdPfLMUpao3eXyTI1IeXM2Tca7ti5O9mCTFFZLHU4LKk4QwImQwEmQXJS1AJ2D5HAwhJy9dm4jHnAEtSuCCnJIcHunrCUGipzVipYNyTxAF1qiTXaP+EvD6V/VVdVV+aPZs8NRd1d19a/qmfcLaGxP1fPU89RTRQH7/fbzPAAAAAAAYCyR5AAAAAAAAAAAAGOJJAcAAAAAAAAAABhLJDkAAAAAAAAAAMBYIskBAAAAAAAAAADGEkkOAAAAAAAAAAAwlkhyAAAAAAAAAACAsZQYdQcAAACAzcIwjNXvvu93XS5M/V7qAgAAAMC4YSYHAAAAMATNJEMzwbA26RC2nO/7gcmRZpn1CY61x9u1DQAAAADjhpkcAAAAwJA0Ew9BiYaw5dZqJjL6pXCpoPylvCQpdyin7GK2ZbmFl6Nd/+rzwXWvPt/+XLu6neoE1Q9bN+r1u2kjav/W1xt2e4Nqs9fx7HS9oGt0O6a9vNft6g/iWfT7HR10vVZ1u60PAAA2JpIcAAAAwAbRbkmq9cmSTsmQ8yfP6/KFy0puSUqGpHtFnYqj/U/t18nXTva93+0svNw6gNkpiNw8FyW42669bo2if1GTTUF9HcQ4dttmr+PZTftRyvVTv8d0EP+GBvGu9VK3X/9uAQDA+CLJAQAAAGwQrZaoWv+91d+SdO2dazpz9IzSu9KaPzKv1HRKhmnIczzZJVvWsqUrb1zRS5Mv6fTbp5U7nJMUHAgNE3zsFIRtF8AMajdMcHft+aB63RpV/5rnug0ot0ssDHIcuwm+Rx3PfujXmAaJOqZR6/U6pr2OS5SkR7fvKQAA2BzYkwMAAADY5K69c03nvnFO2cWs9n59r+aemFPuUE6Zgxnt/tJu7Xxkp2b2zmh236zSe9I6++RZXX/3+kD7FCa42u25TuX6GSAdRf+uPh89qN5NADyoXNilmNqV67Yv/bznVtceReA86rvZTb1exjTquESt14/3FAAAbGwkOQAAAIAhaS4ZFbTPRthy/XLm6BntfHinsotZ5Q7l9OBXH1T28ax2f3m3djy8QzMLM9o2t03TmWltzW5V8oGkXjnyylD61k9hA+yjWKZobbtx69/69uLaz7DiGBiPOqbj/iz6aTPcIwAAaI3lqgAAAIAhaO6L0UxctFs+Kqjc+u/tNikPuyfH+ZPnld6d1uy+We364i5lFjOaWZhR3a6rfKOsxFRChmHIcz3V7bpcy5VTcWStWHr96dd1/PvHex8cbApR9s8YRPujai+OyRUAAICNgCQHAAAAMCTtNvxefzxsuW7Otzt3+cJlzR+ZV3pPWlvntmpmYUbb57fLWrZUt+tyKo7soq3UdKrxSaeUTCc1tWNKH7z2wcCSHKPY3HgY4t6/YepHEqDbTcRHmWiIQx/C4B0FAADjhiQHAAAAsEkVLhWU3JJUajql5JakzKSpul2XtWzJtVz53r1ZJROGzIQpM2nKTJlKpBKNz1RCS+8vKfNYpqd+dAqqRg0Ixz2QHIf+XX2+MfatNm0eZKC73bV7SQJE3eB+UIY5noMSh3dUGt17CgAAxgd7cgAAAAADVigU7vusPf7ZZ5/p4sWLq8eG+f2tV9+SDMkwDfmeL6fsqHyjrGK+qMqtiuySLddy5bmefM+X/HvLZJmSMdGok38vH2VIBiLuv5SPc/+aQeTmZxiaG1EPYkPqVuWGJWiD7DgH5uP8jkqjeU8BAED8MZMDAAAAGKAXX3xRjzzyiI4dO/a5c77vr37WHhvW98YByXM8OSVH1TtVJaYSciqODMOQa7mqrdTklB25lqu6U28kPOp+I+nRJ+0Cqu1+vd2ubKdrjVpc+9duY+pBb1jdLhEQ9pl3m+CIw7g37y+u4jRW643qPQUAAOOBJAcAAAAwYB999JFOnTr1ueO5XE6S7kuADPX7qWP6+Lsfyy7Zqi5XNXlzUoZhyC7aMiYam407ZUe1lZrssi2ncn+ywzAN5Q7ngm4/srBB7zgHZ6X490+Kd9/WizKenYLg4/B8Bm1cxiDu/QMAAKNBkgMAAADYpLKLWTkVR9aypcqtihKTCXmup9R0SmbClO/5ci1XdtlW7W5NduleoqPiyrUan8zB3vbj6FXcg7Nx718ncex7HPs07sZ9TMe9/wAAoHfsyQEAAABsYvuf2t/Yg+NmRaWlkor5YuNTKKq0VFL5ZlmVWxVVP6uqdre2OqvDumvpwNMHRtr3boObQUvb9DtYGvf+RRW1n732O8r9r9/3o9UeIFH3BOlWp9kkvY5p1HcmLu8UAABAL0hyAAAAAJvYyddOyqk4q0mNYr6o4vVGoqO0VFL507IqNyuq3q6qeqeR6LDuWpInHT93fKB9G+QSQ+uv3e81/ePev6DrRt3UO2w/W5Xr1Oa4BOODxlMa3pgG1RuXMZXin/gDAACjxXJVAAAAwCZ3+u3TOvvkWRWvFeVUHCXTSSVSCcmU/LqvulOXW3FXZ3D4vq/n3nmub+0HBWPXBzDXlg/z6/j1x5p1WtXtR7B0FP0LE1xvVz9KAiVqP4PqtdLLePYi6phGTUgNakyDnnm3Y9rvcQnzjnaqDwAAwEwOAAAAYJPLHc7pmR8+o9KtkpY/WV5dsqpUKK3O5ijfLKtYKMopOXr2jWeVWRz8XhyDWkaoU7A4DobZv05tBbUXtZ+9tBl3nfo/qjHdCDbyOwMAAHrHTA4AAAAAmntiTi/UXtCFb13Qhz/4UImphHzPlyQZpiHXcnXg6QOhl6gKE3iMGpzsR1BzkIHRUfSvlzZHUbfbfTUGIWrioNfrDqr+sMZ03MYFAABsfCQ5AAAAAKw68eoJnXj1hJbeX1L+vbykxkyPzMHBz9wAAAAAgG6R5AAAAADwOZnHMso8RmIDAAAAQLyR5AAAAECsGIax+t33/a7LhanfS11sPt1seMySOgAAAMBwsfE4AAAAYqOZZGgmGNYmHcKW830/MDnSLLM+wbH2eLu2AQAAAADxwUwOAAAAxEoz8RCUaAhbbq1mIqPT9bpRuFRQ/tK9fSsO5ZRdzHZ9DcQfszMAAACA+CLJAQAAgE2l05JUYZerOn/yvC5fuKzklqRkSLpX1Kk42v/Ufp187WRf+wwAAAAAaI0kBwAAADaVVktUhTknSdfeuaYzR88ovSut+SPzSk2nZJiGPMeTXbJlLVu68sYVvTT5kk6/fVq5w7nB3xAAAAAAbGIkOQAAAIAQrr1zTee+cU7Zxaxm980qvSet5JakfM+XU3JUXa6qcqui1HRKxUJRZ588q2d++Myouw0AAAAAGxpJDgAAAMRKcwZF0D4bYct12247Z46eUXYxq+xiVru+uEtb57bKTJpyyo6qd6qavDmpxGRC5oQpSSpeK+qVI69I/21fugcAAAAAaIEkBwAAAGKjmbRoJi7aLR8VVG7993ablK9fqqrdufMnzyu9O63ZfbPa9cVdyixmNLMwo7pdV/lGWYmphAzDkOd6qtt1uZYrp+LIWrFkn7cltugAAAAAgIEgyQEAAIBYaTebYv3xsOW6Od/u3OULlzV/ZF7pPWltnduqmYUZbZ/fLmvZUt2uy6k4sou2UtOpxiedUjKd1NSOKdmX7Y79AQAAAABEZ466AwAAAECcFS4VlNySVGo6peSWpMykqbpdl7VsybVc+d69WSUThsyEKTNpykyZSqQSSqQSUkIylvqzpBYAAAAA4H4kOQAAABALjz76qAqFwuqnqVAo6KOPPlr9++LFi0P9nr+UlwzJMI3GJuNlR+UbZRXzRVVuVWSXbLmWK8/15Hu+5N9bJstUI/FhmFI+0pAAAAAAAAKwXBUAAABG7jvf+Y7y+XzL5aJ83//c3hlD/+5LnuPJKTU2GU9MJeRUHBmGIddyVVupySk7ci1XdafeSHjU/UbSAwAAAAAwMCQ5AAAAEAu5XC7U8WPHjg31e2FbY1aJXbJVXa5q8uakDMOQXbRlTDQ2G3fKjmorNdllW05lXbLD9+XnSHYAAAAAwCCQ5AAAAAA6yC5m5VQcWcuWKrcqSkwm5LmeUtMpmQlTvufLtVzZZVu1uzXZpXuJjorb2LPD9aXMqO8CAAAAADYmkhwAAABAgP1P7deVN640EhsTjY3HU9MpmUlT8qW6U5dTcWSXGomO5qwO664l7R917wEAAABg4yLJAQAAAAQ4+dpJvTT5koqFoiTJtVyl0imZKVOG0ViyyrVcOZXGslW1u7VGgsOTdFISq1UBAAAAwECYo+4AAAAAMA5Ov31aTslR8VpRxXxRK/kVlfIlFQtFlZZKKt8sq3Kzouqdqip3KvJ9X8+989youw0AAAAAGxpJDgAAACCE3OGcnvnhMyrdKmn5k2UV841kR6lQaiQ5Pi2rfLOsYqEop+To2TeeVWaRzTgAAAAAYJBYrgoAAAAIae6JOb1Qe0EXvnVBH/7gQyWmEvK9xlpUhmnItVwdePqAjp87K9qQAAAAIABJREFUPuKeAgAAAMDmQJIDAAAA6NKJV0/oxKsntPT+kvLv5SU1ZnpkDjJzAwAAAACGiSQHAAAAEFHmsYwyj5HYAAAAAIBRIckBAAAADIlhGKvffd+PVK7dubXHg853ahsAAAAAxgkbjwMAAABD0EwyNBMMrZISQeUMw5Dv+6uf9YmLtZ/112xXDwAAAADGGTM5AAAAgCFpJh+CEg3tyoWdgdFMavSicKmg/KV7+40cyim7mG1ZbuHlaNe/+nxw3avPB19n/TXC1GlVr5u6Ua/fTRtR+zfs8ehlHLup2+t4drpe0DW6HdNe3+thPYt+v6ODqhf2vzH9/PcLAADGC0kOAAAAYIwMetmp8yfP6/KFy0puSUqGpHtNOBVH+5/ar5Ovnex7m+0svNx90DvMNYPO9Ros7bWNdvVHMR797meUNvv9zMKOVdQx7UW/x7RdvV7GdBDvGgAAQC9IcgAAAABjZP0+G+2WplpfJ2iJqmvvXNOZo2eU3pXW/JF5paZTMkxDnuPJLtmyli1deeOKXpp8SaffPq3c4Zyk4EBomOBzpyBsUHC3eS5MADXo1/th2wwjaFzCBJ/Xnh/EeLSqN6x+tmpz/XWDynbbXlT9GtMgUcc0ar1ex7TXcenHDI1+JSYBAMB4Y08OAAAAYBNot1+H1EhwnPvGOWUXs9r79b2ae2JOuUM5ZQ5mtPtLu7XzkZ2a2Tuj2X2zSu9J6+yTZ3X93esD7W+YAHnUwGa7ev0KlIYJDHd7jUGMR6cA8aD6GRSU7rYv/bznVtceRfC82zGNUq+XMY06LqMaTwAAsPGR5AAAAACGpDmbImhWRbty/dgwvNVMjzNHz2jnwzuVXcwqdyinB7/6oLKPZ7X7y7u14+EdmlmY0ba5bZrOTGtrdquSDyT1ypFXeu4L7hc2ATDsZX/WtxfXfoYVx0B71DEd92fRi414TwAAIBqWqwIAAACGoLlkVDNR0W7ZqU7lgjYib7fheKc650+eV3p3WrP7ZrXri7uUWcxoZmFGdbuu8o2yElMJGYYhz/VUt+tyLVdOxZG1Yun1p1/X8e8fjzok2GRGvbTQqJNDcUyubASMKwAAYCYHAAAAMCTtloxq9Xe7paWCznXTriRdvnBZOx7eofSetLbObdXMwoy2z2/Xll1bNDUzpcltk0pNp376SaeUTCc1tWNKH7z2QTe335VBBKSDftU+jCA8vz7/qYWX7/9EvUY35UYZEO/lPodpHPoIAACwFjM5AAAAgE2qcKmg5JakUtMpJbckZSZN1e26rGVLruXK9+7NKpkwZCZMmUlTZspUIpVofKYSWnp/SZnHMj31o1NQtd9B6avPdw42DysIHodfn68di3Ybjw/CIJJMUTe4H5RhjuegxOEdbScOSSsAABAfzOQAAAAABuzRRx9VoVBY/TStPRZ0/C//8i9Xz1+8eLEv39969S3JkAzTkO/5csqOyjfKKuaLqtyqyC7Zci1XnuvJ93zJv7f0lSkZE406+ffyPY7O5hHnwGw/ZlV0q7kRddQNqcOO57ATDEGbp8c54RHndxQAAKAdZnIAAAAAA/Sd73xH+Xy+6+WlOh3r1/fGAclzPDklR9U7VSWmEnIqjgzDkGu5qq3U5JQduZarulNvJDzqfiPp0SftAqrtZhn0olMQdxDthWl3lNot4TXoDavbJQLCPoNuExxxGPfm/cVVnMYKAACgGyQ5AAAAgAHL5XI9H//KV76y+v3YsWP9+X7qmD7+7seyS7aqy1VN3pyUYRiyi7aMicZm407ZUW2lJrtsy6ncn+wwTEO5w63voR+6CXqHERTE7Xd7YduNgzj3bb0o49kpuTAOz2fQxmkMxqmvAABgOEhyAAAAAJtUdjErp+LIWrZUuVVRYjIhz/WUmk7JTJjyPV+u5cou26rdrcku3Ut0VFy5VuOTOdjbfhwb3TgHZOPY9zj2adwxpgAAYNyxJwcAAACwie1/an9jD46bFZWWSirmi41PoajSUknlm2VVblVU/ayq2t3a6qwO666lA08fGHX3Y63b4HHQElFxCUZH7Wev/Y5y/+v3/Wi1B0jUPUG61Wk2Sa9jGvWdics7FVacl/sCAACjQ5IDAAAA2MROvnZSTsVZTWoU80UVrzcSHaWlksqfllW5WVH1dlXVO41Eh3XXkjzp+LnjA+3boAKaQQHhfrYRNXi8vi+jGotuN/UO289W5YL2SwnTn1EL824Na0yD6o3LmLYyjn0GAACDw3JVAAAAwCZ3+u3TOvvkWRWvFeVUHCXTSSVSCcmU/LqvulOXW3FXZ3D4vq/n3nmub+0HBWM7Bb2Djq//xX7zfJhf1Ue19trdthPUx6Bf5Acd72YsO4naz7DPoF3/BvncOrXb6fj6dqMmpAY1pkHPvNsx7fe4hHlHAQAAOmEmBwAAALDJ5Q7n9MwPn1HpVknLnyyvLllVKpRWZ3OUb5ZVLBTllBw9+8azyiwOfi+OQSwj1Omaw1q2KEin/g2zraD2ovYz7uPfi079H9WYbhTjPPMEAAAMFjM5AAAAAGjuiTm9UHtBF751QR/+4EMlphLyPV+SZJiGXMvVgacPhF6iKkwgspdgZa+BzkEGSvtx7W6vMaqxjFq32301BiFq4qDX6w6q/rDGdNzGBQAAbHwkOQAAAACsOvHqCZ149YSW3l9S/r28pMZMj8zBwc/cAAAAAIBukeQAAAAA8DmZxzLKPEZiAwAAAEC8keQAAAAAgA662UiaJXUAAACA4WLjcQAAAAAAAAAAMJaYyQEAAAAAHTA7AwAAAIgvZnIAAAAAAAAAAICxRJIDAAAAAAAAAACMJZIcAAAAAAAAAABgLJHkAAAAAAAAAAAAY4kkBwAAAAAAAAAAGEskOQAAAAAAAAAAwFgiyQEAAAAAAAAAAMYSSQ4AAAAAAAAAADCWSHIAAAAAAAAAAICxRJIDAAAAAAAAAACMJZIcAAAAAAAAAABgLJHkAAAAAAAAAAAAY4kkBwAAAAAAAAAAGEskOQAAAAAAAAAAwFgiyQEAAAAAAAAAAMYSSQ4AAAAAAAAAADCWSHIAAAAAAAAAAICxRJIDAAAAAAAAAACMpcSoOwAAAAAAo2YYxup33/cjlRvEOQAAAACdMZMDAAAAwKbWTDI0Ewxrkw5hyxmGId/3Vz/9OAcAAAAgGDM5AAAAAGx6zcRFUKKhXblOMzCinmuncKmg/KW8JCl3KKfsYrZluYWXu760JOnq88F1rz4f7lqtrhO27vr63dQLe821Bn1P6+sNu71BtdnreHa6XtA1uh3TXt/rYT2Lfr+jUf/Nhanf7+cPAEAUJDkAAAAAYMD6sSTV+ZPndfnCZSW3JCVD0r3LOBVH+5/ar5OvnexDT8NZeDla4DNM3TDXiCqoX1K0++p0T1Hvo5cxjNLPKG32Op7dtB+lXD/1e0zb1etlTHsZlyjvW7+fPwAAUZHkAAAAAIA+ai5Btdbav1ud73T82jvXdOboGaV3pTV/ZF6p6ZQM05DneLJLtqxlS1feuKKXJl/S6bdPK3c4Jyk4EBom+NgpsBkmWdFtYHQYgsYlTEB37fmw49E81+39t0ssDKqfrdpcf92gst22F1W/xjRI1DGNWq/XMY06Lu3ajjIDZhjPHwCAJvbkAAAAAIA+aZeoiFrv2jvXdO4b55RdzGrv1/dq7ok55Q7llDmY0e4v7dbOR3ZqZu+MZvfNKr0nrbNPntX1d6/341baCrukT7ty3czi6HdwNExguNtrhEkYRA2qdxMADyo3iOfWj/EM2/76a48icN7tmEap18uYRh2XqP9mO7VHYgMAMEwkOQAAAABses3lpII2/u5UrtMMjaBrtkuMnDl6Rjsf3qnsYla5Qzk9+NUHlX08q91f3q0dD+/QzMKMts1t03RmWluzW5V8IKlXjrzSsb1xEacgadgg8LBnqaxvL679DCtOz7wp6piO+7MAAGCcsFwVAAAAgE2tuYl4MxnRbmmpoHJr/3ft+U6blHeqd/7keaV3pzW7b1a7vrhLmcWMZhZmVLfrKt8oKzGVkGEY8lxPdbsu13LlVBxZK5Zef/p1Hf/+8T6MTnf6MQODoO/wjXr/hFEnh+KYXBmWUT97AAD6gSQHAAAAgE2v3UyKTntrhDneS73LFy5r/si80nvS2jq3VTMLM9o+v13WsqW6XZdTcWQXbaWmU41POqVkOqmpHVP64LUPBpbk6CYg3W0weZQBV5IrP9WPJEC3m4iPMsgehz6EMYx3tF8JoHEZUwDAxkCSAwAAAABipnCpoOSWpFLTKSW3JGUmTdXtuqxlS67lyvfuzSqZMGQmTJlJU2bKVCKVaHymElp6f0mZxzI99aNTULVT8LJdvTCBz1EHRUfdfrMPCy+33rR5kIHuXp5bO1E3uB+UYY7noAxivHp99q3qx+HfEgBgc2BPDgAAAACbVqFQuO/T6fjFixdXzw/6+1uvviUZkmEa8j1fTtlR+UZZxXxRlVsV2SVbruXKcz35ni/595a8MiVjolEn/14+6rD0TXNj4jAbIo8y2BznX503kx3NzzB089xa6SUwPkhBm6fHOeExrHe012e/VpzHEwCwsZDkAAAAALApvfjii3rzzTfl+/7qp2ntsbV7a6w9P8jvjQOS53hySo6qd6oqLZVUzBdV/rSs6u2qais1OWVHruWq7tQbCY+630h69Mn6gOfawGdQALNTQLnbTZoHKa4JjnZB5n4En4PabXcsTNC62wRHHMY9Dn3oZJgJjnbHwvx7j/LfCQAA+oHlqgAAAABsWh999JFOnTr1ueO5XO5zx44dOza876eO6ePvfiy7ZKu6XNXkzUkZhiG7aMuYaGw27pQd1VZqssu2nMr9yQ7DNJQ7/Pl76JdOyyn1olNAdBCB3jgF2tuJc9/WizKew37m42acx2BQ/50AAGA9khwAAAAAEDPZxayciiNr2VLlVkWJyYQ811NqOiUzYcr3fLmWK7tsq3a3Jrt0L9FRceVajU/mYG/7cWx04xw8jmPf49incceYAgAQDkkOAAAAAIih/U/t15U3rjQSGxONjcdT0ymZSVPypbpTl1NxZJcaiY7mrA7rrqUDTx8YSZ+bv9yOUq+dOMzgCPpFelyC0VH7GfW5BV23k2E/80463XuvYxr1nRnmElUsKQUAGHfsyQEAAAAAMXTytZNyKo6KheLqfhzF60UV842/y5+WVblZUfV2VdU7VdXu1mTdtSRPOn7u+ED71s3eDK2OjTIZ0Gsf2u0n0m/trht1U++w/ez2ucXhmYYRNJ7S8MY0qN6oxjTqs+/megAADAIzOQAAAAAgpk6/fVpnnzyr4rWinIqjZDqpRCohmZJf91V36nIr7uoMDt/39dw7z/Wt/aibizfrxS3IubY/Qb/eb3Ws030F/SI/6HiUIHIrUfsZ5bn1Mp69iDqmUd/HQY1p0DPvdkyjjksv/2bDlI97AgwAMP6YyQEAAAAAMZU7nNMzP3xGpVslLX+y3JjNkS+qVCitzuYo3yyrWCjKKTl69o1nlVkc/F4cV5/vHLjsFPQd54Bnp/saZltB7UXt50Z9blLw+zqKMY2TKM8+zL3H7T4BABsTMzkAAAAAIMbmnpjTC7UXdOFbF/ThDz5UYioh3/MlSYZpyLVcHXj6QOglqsIEHfsRmOxXcLOfQdJR3FcvbY6ibr/21ehF1MRBr9cdVP1hjeko7o8kBgAgDkhyAAAAAMAYOPHqCZ149YSW3l9S/r28pMZMj8zBwc/cAAAAAOKKJAcAAAAAjJHMYxllHiOxAQAAAEgkOQAAAAAAY6qbTZJZVgcAAGBjYuNxAAAAAAAAAAAwlpjJAQAAAAAYS8zOAAAAADM5AAAAAAAAAADAWCLJAQAAAAAAAAAAxhJJDgAAAAAAAAAAMJZIcgAAAAAAAAAAgLFEkgMAAAAAAAAAAIwlkhwAAAAAAAAAAGAskeQAAAAAAAAAAABjiSQHAAAAAAAAAAAYSyQ5AAAAAAAAAADAWCLJAQAAAAAAAAAAxhJJDgAAAAAAAAAAMJZIcgAAAAAAAAAAgLFEkgMAAAAAAAAAAIwlkhwAAAAAAAAAAGAskeQAAAAAAAAAAABjiSQHAAAAAAAAAAAYSyQ5AAAAAAAAAADAWCLJAQAAAAAAAAAAxlJi1B0AAAAAAAyOYRir333f77pcN/Xb1QuqCwAAAETFTA4AAAAA2KCaiYZmgmF94iFMOd/3AxMU7a7brEuCAwAAAIPCTA4AAAAA2MCaCQbf99smI7opt15zBkc3ddopXCoofykvScodyim7mG1ZbuHlaNe/+nxw3avPB1+n1TWi1AtTpxvt7i1sO8O+r361N6g2ex3PTtcLuka3Y9rrez2sZ9Hvd3TQ7xoAYDyQ5AAAAAAARLJ+iapW55s6lTt/8rwuX7is5JakZEi6V9SpONr/1H6dfO1kv7ocaOHlaEHaTvWiJmW60amN5rm43FdQX/vdzyht9jqe3bQfpVw/9XtM29XrZUx7GZeo9wcAGB8kOQAAAAAAXQtKcKw/16r8tXeu6czRM0rvSmv+yLxS0ykZpiHP8WSXbFnLlq68cUUvTb6k02+fVu5wTlJwIDRM4LJTEDZMkHbt+aB66+sMKpAdNC5xu692iYVB9bNVm+uvG1S22/ai6teYBok6plHr9Tqm3Y5LP94ZAED8sScHAAAAACASwzBWP82/w7r2zjWd+8Y5ZRez2vv1vZp7Yk65QzllDma0+0u7tfORnZrZO6PZfbNK70nr7JNndf3d64O6FUnhg53ry4UJrA86kBomMNztNQZxX50SUYPqZ1Dyq9u+9POeW117FEH3bsc0Sr1exrTXcYl6fwCA8UCSAwAAAAA2sLAJiG4TFWs3FV+7n0dYZ46e0c6Hdyq7mFXuUE4PfvVBZR/PaveXd2vHwzs0szCjbXPbNJ2Z1tbsViUfSOqVI6+Evn6/hQ2Uj2K5oV7E9b7WtxfXfoYVx6B61DEdl2cxLv0EAPSO5aoAAAAAYINqbgjeTFysTUKsXT4qqNz672GSGeuTJWvrnD95Xundac3um9WuL+5SZjGjmYUZ1e26yjfKSkwlZBiGPNdT3a7LtVw5FUfWiqXXn35dx79/vNuhwCYVZf+MQbQ/qvbimFwBAKDfSHIAAAAAwAbWLiGx/njYcmHb6VTv8oXLmj8yr/SetLbObdXMwoy2z2+XtWypbtflVBzZRVup6VTjk04pmU5qaseUPnjtg4ElOTbqL7o36n1F0Y8kQLf7QYwy0RCHPoTBOwoA6AVJDgAAAADA0BQuFZTcklRqOqXklqTMpKm6XZe1bMm1XPnevVklE4bMhCkzacpMmUqkEo3PVEJL7y8p81imp350CqrGPSAcVRzu6+rzjbFvteHzIAPd7a7dSxIg6gb3gzLM8RyUOLyjAIDxw54cAAAAALABFQqF+z5Nd+/e1cWLF1f/Hvb3/KW8ZEiGacj3fDllR+UbZRXzRVVuVWSXbLmWK8/15Hu+5N9b+sqUjIlGnfx7+UhjshnF+Zf8zWRH8zMMzQ2so25kHXY8h51gCNo8Pc4Jjzi/owCA8cBMDgAAAADYYF588UU98sgjOnbs2OfOrd0ovPn30L/7kud4ckqOqneqSkwl5FQcGYYh13JVW6nJKTtyLVd1p95IeNT9RtKjT9oFVNvNMhhHcQ0et9vwedAbQbdLBIR95t0mOOIw7s37i6s4jRUAYHyR5AAAAACADeijjz7SqVOnPnd8ZmbmvuTHsL8XtjVmldglW9XlqiZvTsowDNlFW8ZEY7Nxp+yotlKTXbblVO5PdhimodzhXNDtR9ZN0DvOxiF4HOe+rRdlPDslF8bh+QwaYwAA6BeSHAAAAABCMQxj9XunTaXblQuq3+l82LYRf9nFrJyKI2vZUuVWRYnJhDzXU2o6JTNhyvd8uZYru2yrdrcmu3Qv0VFx5VqNT+Zgb/txbHTjHDyOY9/j2Kdxx5gCAPqJPTkAAAAABGomGZoJhrVJh7Dl1i+TtL5e87zv+/fV63QO42n/U/sbe3DcrKi0VFIxX2x8CkWVlkoq3yyrcqui6mdV1e7WVmd1WHctHXj6wEj6HLSUUlyCtt32Y1zuK2o/e+13lPtfv+9Hqz1Aou4J0q1Os0l6HdOo78yw3qlxebcBAL0jyQEAAAAglGaCImgmRdhy3bbbjertqq786yu68q+vqHqn2pd+oH9OvnZSTsVZTWoU80UVrzcSHaWlksqfllW5WVH1dlXVO41Eh3XXkjzp+LnjA+1bmP0L1peJy54HvQZth3VfvQado/azVblObY5LEDxoPKXhjWlQvVGNaVz/zQIA+oPlqgAAAABsGD/6r3+k9/7pe7LLtpJTSUmSW3WVnE7q0H98SF/7H7424h6i6fTbp3X2ybMqXivKqThKppNKpBKSKfl1X3WnLrfirs7g8H1fz73zXN/aDwpydtqkul39oF+uBx3vJfC79lphfr2//tiw7ytKkDlqP4PqtdLLePYi6phGDdoPakyDnnm3Yxp1XKLeHwBgvDCTAwAAAMDINZehan7aaS5dtd7KX6/oH/3MP9Kf/a9/ppn5GS380oLmfmFO2cWs9hzcowdmHtBP/vAn+sfZf6zi9eIgbwUh5Q7n9MwPn1HpVknLnyyvLllVKpRWZ3OUb5ZVLBTllBw9+8azyiwOfi+OoGWEOgV9x9kw76tTW0HtRe1nL23GXdD7OooxjZNx6ScAIDpmcgAAAACIhXablK891irBsfzJsr73C99Tendau/fv1nRmWsktSfmev7rBdXVnVVM3p7RyfUV/vPjH+q2f/NZA7wXhzD0xpxdqL+jCty7owx98qMRUQr53bz8X05BruTrw9IHQS1SFCVr2I7DZ7TWGEUwdt/saRd1u99UYhKiJg16vO6j6wxrTUd0fAGA8kOQAAAAAEEozyRC08XfYckH1g441/dHBP9LM/Ixyh3Pa9cVdms5OayI1IbfqqvpZVZWbFSWnkjKTpmRIK3+zoj9+7I+l/yxS9zAAJ149oROvntDS+0vKv5eX1JjpkTk4+JkbAAAAGG8kOQAAAAAEWrucVPPvprUJiKBy6783z6891yrB0a7Mv/gP/4UmEhPavX+3dn95tzIHM9o+v111t67qraqSS0kZpiHfu7fHg+XKtVwtf7Is/R+S/v3exgX9lXkso8xjJDYAAAAQHkkOAAAAAKG0m0mx/njYcv049+f/7M8198ScprPT2vbgNu34uR2a+cKMrM+sxnJVVUd22ZZdspXamlJqOqVUOqUHZh6Q82cOSQ7EVjcbSbMUDwAA2MzYeBwAAADAWLrx4Q0lUglNbZ9Sajq1up+DXbRVd+oyZMicMGUmTE0kJzSRmtDE5L3P1ISMCUO6Oeq7AAAAANALZnIAAAAA6Oj27dv3/T07O3vf8UQioU8++UQ///M/L0n6i7/4i6F8L1wqSKYayQpfcqqOKjcr8hxPnufJLtlybVd+3b9vWSzDbCQ/DMOQkY+2bwgwaMzOAAAACIeZHAAAAADaevnll/XjH/9YxWJx9dPU/LtSqejjjz9ePT6s73bFliFDnuvJLtuqflZVaamklfyKyp+WVf2sKnvFllNxVK/VG8mPuiffayQ9DBny7fZLYQEAAACIP2ZyAAAAAGhreXlZ7777rn7913/9c+e+8IUvrH7/jd/4jaF/zx7Myvd9OWVH1rKlyo2KDMNQqpiSmTBVd+pyyo5qKzXZJVtO1VHdqsuzPXmup7pfl5FjJgcAAAAwzkhyAAAAABhLuUM5OVYjwVG9XVViKiGv7ik13Uhy+J4v13Jll+xGoqNoy67YcqxGskOO5OeYyQEAAACMM5IcAAAAAMaSmTT18K89rPxP8o3ExoQpz/aUnE5qIjkh3/dVt+tyq65qxdpqoqM5u0P7JDGRAwAAABhrJDkAAAAAjK1v/m/f1O+lf0/lG2VJkmu5q0kOGZLnenItt5HYKNZUu3sv2VGxpadH3HkAAAAAPWPjcQAAAABjKzGV0FPff0rV21WVCiUV88XGp1BUqVBSeams8o2yKrcqqt6uqrpclWu5+ubr3+T/DQEAAAAbADM5AAAAAIy1Lz31JRkThs4/fV61lZomt09qYnJChmlInlR363ItV7WVmpyqo29e+KYe/buPSn8x6p4DAAAA6BVJDgAAAABj74vHv6jf+fR3dO7Xz2npz5fk+74Mw5AvX/IkwzSUWczo7/3Lv6fJbZOj7i4AAACAPiHJAQAAAGBDeGDHA3ruT5/TyrUVXf/xdV39v69KkhaOLmjuiTlte3DbiHsIAAAAoN9IcgAAAADYULY9uE3bHtymL5340qi7AgAAAGDA2GoPAAAAAAAAAACMJZIcAAAAAAAAAABgLJHkAAAAAAAAAAAAY4kkBwAAAAAAAAAAGEskOQAAAAAAAAAAwFgiyQEAAAAAAAAAAMYSSQ4AAAAAAAAAADCWSHIAAAAAAAAAAICxlBh1BwAAAAAAwP0Mw1j97vt+T+UMw/jcuXb11h4PahsAACAOmMkBAAAAAECMNBMNzQTD+sRDN+XaHfN9f/XTKrHR/AAAAMQdMzkAAAAAAIiZZoKhVRIibLlmMqNT/X4oXCoofykvScodyim7mG1ZbuHlaNe/+nxw3avPtz/Xrm6UOt1cI6wo/QuqH6bu+nrDbm9QbfY6np2uF3SNbse0l/e6Xf1BPIt+v6NRx6XbZ9jLOwdgvJDkAAAAAABgg2m1RFU3dZs6XeP8yfO6fOGykluSkiHpXlGn4mj/U/t18rWTkdqPYuHl1sHLTkHk5rlRBj177V+7+u3GI6jNToL62u9+Rmmz38877FhFHdNe9HtMB/FvKMq49OMZ9vKuAhhPJDkAAAAAANhAghIcnWZ3tNq7Y/2xa+9c05mjZ5Teldb8kXmlplMyTEOe48ku2bKWLV1544pemnxJp98+rdzhnKTgQGiYwGOnIGy74GVQu63qhQkS9yt3S65HAAAgAElEQVRQGqV/a8+vv0ZQvfV1ug1Et0ssDKqfrdpcf92gst22F1W/xjRI1DGNWq/XMe12XPr1DLtN2gAYX+zJAQAAAADABmMYxuqn+fdaUffduPbONZ37xjllF7Pa+/W9mntiTrlDOWUOZrT7S7u185Gdmtk7o9l9s0rvSevsk2d1/d3rfbuvVsIEV7s9Nyz96F83CZrm+ahB9W4C4EHlwv4av125bvvSz3tude1RvE/djmmUer2MaZRx6fUZRnlvAIw/khwAAAAAAMRMu+REmHJrExhr9+xoV7+bRMeZo2e08+Gdyi5mlTuU04NffVDZx7Pa/eXd2vHwDs0szGjb3DZNZ6a1NbtVyQeSeuXIK6GvH3dx+RV42EDusPu7vr249jOsOAbEo47puD8LAOiE5aoAAAAAAIiR5nJSzcTF2iTE2qREp3KddNpzo9WMj6bzJ88rvTut2X2z2vXFXcosZjSzMKO6XVf5RlmJqYQMw5DneqrbdbmWK6fiyFqx9PrTr+v49493MQrxFsfg90Yy6v1SRp0c4v2KZtTvDYDRIckBAAAAAEDMtEtYrD8eJrHRTZ1O5y5fuKz5I/NK70lr69xWzSzMaPv8dlnLlup2XU7FkV20lZpONT7plJLppKZ2TOmD1z4YWJJjEBtpx0Hc+zdM/UgCdLuJeBw2pI97sH7UyaBuy8d9PAFER5IDAAAAAAB0VLhUUHJLUqnplJJbkjKTpup2XdayJddy5Xv3ZpVMGDITpsykKTNlKpFKND5TCS29v6TMY5me+tEpyBk1gBm23qgCz3EIzF59vnH/rTZ8HmSgu921e3kWUTesHpRhjuegxO3fxCDeGwDxRpIDAAAAAICYKBQK9/2dzWb7dvzixYs6evSoJHX9/a1X35IMyTAN+Z4vp+yofKOsul2X7/myS7Zcy5XnevI9X/LvLX1lSsZEo07+vXzPSY5+iXuwM879G0UQvtdEQNjxHPa9tds8vdmPVkmluBj2OxqlvY2QQAIQDhuPAwAAAAAQAy+++KLefPPNz20aLrXeTLzb4+vPd/O9cUDyHE9OyVH1TlWlpZKK+aLKn5ZVvV1VbaUmp+zItVzVnXoj4VH3G0mPPrn6fOuPNF7LEXUS1/6tHeswx/vZbrtjYZ55twmOOIx7HPrQyTgmONYeI9kBbDzM5AAAAAAAICY++ugjnTp16nPHc7lcy/LdHD927Fj076eO6ePvfiy7ZKu6XNXkzUkZhiG7aMuYaGw27pQd1VZqssu2nMr9yQ7DNJQ73Lqv/dBpOaW1ogZnhxXUjVOgvZ049229KOPZKQA+Ds9n0MYhwQFg8yHJAQAAAAAAOsouZuVUHFnLliq3KkpMJuS5nlLTKZkJU77ny7Vc2WVbtbs12aV7iY6KK9dqfDIHR7tUVdyDpXHvXydx7Hsc+zTuSHAAiCuWqwIAAAAAAIH2P7VfxXxRlZuV1aWqivmiioWiSksllW+WVblVUfWzqmp3a6uzOqy7lg48fWCkfe8lWDqMpW267V/QsjtxCQ5H7Wev/Y66vFGn5dDWlhm0Tu9cr2Ma9Z0ZhwTHqN93AKNDkgMAAAAAAAQ6+dpJORVnNalRzBdVvN5IdJSWSip/WlblZkXV21VV7zQSHdZdS/Kk4+eOD7Rvw1hiaFAB1F77t/7eB5WU6TWhErWfrcp1ajMuCZ4gQeMpDW9Mg+qNQ4Kj3TX6fV0A8cRyVQAAAAAAIJTTb5/W2SfPqnitKKfiKJlOKpFKSKbk133Vnbrcirs6g8P3fT33znN9az8oGLs+eLm2fJhfxw9bL/1r7kPSrm7QL/KDjndKIHQjaj+D6rUyqucddUyjJqQGNaZBz7zbMY0yLr0+wyjvDYDxx0wOAAAAAAAQSu5wTs/88BmVbpW0/Mny6pJVpUJpdTZH+WZZxUJRTsnRs288q8zi4PfiGNQyQnH/5XenQO8w2wpqL2o/e2kz7jr1f1RjulFs5PcGQGvM5AAAAAAAAKHNPTGnF2ov6MK3LujDH3yoxFRCvudLkgzTkGu5OvD0gdBLVIUJOkYNTMZ1iap+Xr/ba/TS5ijqxmFPhqiJg16vO6j6wxrTKHX79QxJZgCbC0kOAAAAAADQtROvntCJV09o6f0l5d/LS2rM9MgcHPzMDQAAgCaSHAAAAAAAILLMYxllHiOxAQAARoMkBwAAAAAAQB90s9Exy+kAANAfbDwOAAAAAAAAAADGEjM5AAAAAAAA+oDZGQAADB8zOQAAAAAAAAAAwFgiyQEAAAAAAAAAAMYSy1UBAAAA2PQMw1j97vt+1+WC6nc6H7ZtAAAAAJ/HTA4AAAAAm1ozydBMMKxNOoQt5/t+2wSFYRir533fv69ep3MAAAAAgpHkAAAAALDpNRMUQTMpwpYbtNsf3dbtj26PtA8AAABAHLBcFQAAAAAM0PoZGlETJO//0/f1p7//p7r9/91WItX4v3Ku7Wr24Vn97d/52zp4+mBf+gsAAACME5IcAAAAADBAzSWpWv0dZokqt+bqe7/wPd39m7t6YMcDmjs0JzNlynM82SVb5Ztl/av/4l/p3T94V7/17m/JmGDJKwAAAGweJDkAAAAAYITabWAuSbViTX/w6B9oIjWhhV9a0HRmWsl0Ur7ny626spYtpXenVb5V1mcff6bfn/99/fZHvz3sWxiZXjeMH9Q5AAAADA9JDgAAAACbXnN2RdCsirDleunDWt/9yneVTCf14Fcf1Oyjs9qa26qJ1IRcq5HgqNyqKLklKTNlyjAM3f2bu/qjA38kPdv37sXO2o3gDcNoOX5B5TrNsol6DgAAAMNFkgMAAADAprY2+N38u6nV0lLtyq3/3m5Jqnb11gfJ/+S/+xPVVmp66OsPaff+3cr8fEbbfnabfM9X9XZVpRslmQlT8iXP9VS363Jrru5evSu9JeloL6MyHsIu+xXmWQAAAGA8keQAAAAAsOm1C3avPx62XNTrN3mup/f+6D3N7pvV1rmtmlmY0eyjs9rx0A5V71RlmEYjqVFx5ZQd2UVbtWJNqemUJrdPynnXkfHL7M0RRpSkRzfJks+ufKaf/MFP9PGbH+vOlTuSpJ0P79Tef3evnvhPn9DMF2Yi9hwAAAASSQ4AAAAAiJ3P/uozuRVXUzumNLl1UsktSRmGIafiyPd8GaYhM2nKTJqaSE1oYnJCicmEEpMJJR9ISrck/zNmKYQRZVP4sMtVvfFfvaE/+96fSb40NTOlHV/YIa/uqfJpRZf+l0t6/5X3deg/OaSv/d7X+nxXAAAAm4c56g4AAAAAwKjMzs6qWCyufpqaf1uWpatXr64eH9b3wqWCZEgTyQlJkltzVbldUTFfVPV2VU7ZUd2uy/f8RnDdkAzTkDFhyEyYMg1TykcbE/yU7/urn27981/953r/e+8rczCjh//Ow3roaw9p7ok5ZR7LKPN4Rpmfz2h6z7R+/D//WOf+vXMD6D0AAMDmQJIDAAAAwKb07W9/W0ePHlWhUFj9NDX/Xl5e1ptvvrl6fFjfK7cqMkxDnuvJqTqyPrNUWiqpmC+qfKOs6mdV2UVbbtVV3a7Lczx5dU/yGoF5U6aMysZfrqo50yLMhvGtyoXdQL7bjcXPPnlWS3+xpIVjC5o7PKe5X5hT9lBWP/O3fka7Ht2lHQ/t0PaF7dr6YGMpsqtvX9W5/4BEBwAAQBQsVwUAAABgU1pYWGh7bt++favfv/3tbw/9+56/tUe+58su26ot11S+WZZhGrJLtswJU3W73ji3UpNTduRajWRH3anLd33V/br8PRt7uap+bBgfdVP4TvX+zYV/o2v/7zXNH5nXz3zlZ7TnwB5te3CbZEjWsqXyjbISUwkZpiHf8+U5jU3jP3nrE2mbpH0CAABAF0hyAAAAAEDMZB/Pqm7XVbtbU+V2RYkHEvI9X6liSmbClF/35VquasWaais12SVbTqWR7HBrrvy6L2Nu48/k6HXD+G6uEfb8m//9m9rxhR3aPr9dO39up3Z/Zbdm982qeruqYr4o3/NVt+tyqo6ciiO7bMsu2aqla3J+5JDkAAAA6BJJDgAAAACImdR0Sj/77/ysbv/b25rcOikzYaru1JVKp2QmTclXI1BecRqJjrs12UV7NWCueclPbOyZHHFUKpR058odLRxZ0AM7HtDkzKRS6ZQkyUyYjQ3ipxKf/zyQUDKdlP5aMsobPzkFAADQTyQ5AAAAACCGfvPsb+qf7PsnKt0oSUZj8/FUOqWJVGMzcs/15Fru6rJVzUSHa7nSiRF3fpPKX8qvJjOMicaeKtayJRk/fV6e60l+YzkswzRkTpiNzeITpowJQ36e5BQAAEA32HgcAAAAAGJo+/x2/fJ3flmlQknFQlHF/JpPoajSUknlG2VVblVUvVNVdbmqWrGmX/mHvyJtHXXvB+fixYu6dOnSfZ+mbo+/+uqrq99ffPHFnr8v/9WyzAmzsSRVrbHcWHPD+MqNimrLa/ZQcery6758z19NekwYE9KdXkco/pp7pITZML5duXbn1h7vph4AABhfzOQAAAAAgJg68t8cUXIqqT/5B38iu2hrcutkY5aA2dhY23PuzeYo2nIsR7/2P/2aDv/2Yb3x4huj7vrAvPXWW/J9X7/7u7/7uXOPP/54yzrtjn/rW99a/f6d73yn5+8zD800ZmxUXdVWaqrcqsicMO/fML5k37eHSt2py3M9ea6nuleXdgYMwJhbuwF8M9nQan+TTuXW11n/d7v9UoLqAQCA8USSAwAAAABi7Kv/+Vc1/4vzOv/N8yotlVR365pINpasqtuN7+ldaf39/+vvK7OYGXFvN7fc4zl5dU+1lZqqd6pKbkl+bsN4p+o09k+5t4eKW3EbyQ67Lt/z5ec2ftC9mVhoJjC6LRc1MRGlXuFSQflLeUlS7lBO2cVsy3ILL0fqkq4+H1z36vPtz7Wr26lOmGtFqR90zbXCXr9V/TB119cbdnuDarOfz3v99YKu0e2Y9vJet6s/iGfR73c06rhEvbewdYGNjiQHAAAAAMRc7omcnv+r5/XX/89f6+Mffay/eedvJEk/+4s/q72/slfzvzQ/4h5Ckqaz05p9eFbVW1WVt5d/umH8dEoTyYnGMlZ2vbFB/Mq9z71ZHU7VkWYlpUd9F+OvU0IkbLLk/MnzunzhspJbkpIh6V5Rp+Jo/1P7dfK1k33tcycLL7cOYnYKIjfP9RJo71Wv/WtXv914BLXZSVBf+93PKG3283kHXS9KuX7q95gO4t9QlHEZVHtRnj+w0ZDkAAAAAIBxYEjzvzRPQiPmfuX3fkUXTl1QcmtShmE0Noy/l+SQJM/x5FiO7FJjw3hrxZJdsuVWXek3R9z5MRS0VFWn862Wq7r2zjWdOXpG6V1pzR+ZV2o6JcM05Dme7JIta9nSlTeu6KXJl3T67dPKHc5JCg5Mhgk+dgrCtgvSBrUbFBQetKj9azfDIOx9Nc91G4hul1gYVD9btbn+ukFlu20vqn6NaZCoYxq1Xq9j2u249Ku9KHWBjYyNxwEAAAAA6JNH/+6j+sLRL+ju1bs/3TD++poN4z8tqXKzourtqqzPLNWWa6rcruihrz8kPTLq3o+Xfu+pce2dazr3jXPKLma19+t7NffEnHKHcsoczGj3l3Zr5yM7NbN3RrP7ZpXek9bZJ8/q+rvX+9Z+K90GO8OcW29QvwLvR//WlwuTMIgaVO8mAB5ULuySRe3KdduXft5zq2uPInDe7ZhGqdfLmEYZl0G1R2IDIMkBAAAAAEBfnfo/T2n+l+Z196/vqpQvNRIc+aJKhZJKn5ZU/rSs8s2yKrcqKt8q6+e+9nN6+n9/etTdHprmklGd9uMIKhclwRHU3pmjZ7Tz4Z3KLmaVO5TTg199UNnHs9r95d3a8fAOzSzMaNvcNk1nprU1u1XJB5J65cgrXfUhruIUJA2bABj2Mkrr24trP8OK0zNvijqm4/4sAPSO5aoAAAAAAOizZ/7lM/rRP/iR3vvD91T6tKSJyQklJhv/F9y1XNVrdU08MKFf/C9/UcdePDbazg5RcxPxZsKh3fJRQeXW/u/a8+sTGeuXqmq3J8f5k+eV3p3W7L5Z7friLmUWM5pZmFHdrqt8o6zEVEKGYchzPdXtulzLlVNxZK1Yev3p13X8+8d7G5gRIeg7fKPeP2HUyaE4JlcAjD+SHAAAAAAADMDX/uHXdOg/OqR3//BdffKjT3T7396WJO3ct1MPfe0hPfHbT2jbg9tG3MvhazcDY/3xsOXCnut0/vKFy5o/Mq/0nrS2zm3VzMKMts9vl7VsqW7X5VQc2UVbqelU45NOKZlOamrHlD547YOBJTkGsZH2+jKjCDqTXPmpfiQBut1EfJSJhjj0IYxRJ4Oi1I37mAKDRJIDAAAAAIAB2b6wXb/6P/7qqLuBDgqXCkpuSSo1nVJyS1Jm0lTdrstatuRarnzv3qySCUNmwpSZNGWmTCVSicZnKqGl95eUeSzTUz86BTmjBi+jbHo9TKNuv9mHhZdbb9o8yEB3u2v3ErCOusH9oAxzPAdl2O9omPZajWMc/i0Bo8SeHAAAAAAAYOAKhcJ9n6DjFy9eHMr3/KW8ZEiGacj3fDllR+UbZRXzRVVuVWSXbLmWK8/15Hu+5N9bFsuUjIlGnfx7+cjj0m9hguSjDDbH+VfnzWRH8zMMzQ2lo27wHXY8h/3MgzZPj3PCY9jvaD/ai/N4AsPATA4AAAAAADBQL774oh555BEdO3bsc+fCLEs16O/yJc/x5JQcVe9UlZhKyKk4MgxDruWqtlKTU3Ya+6k49UbCo+43kh590i7A2W6WQbuyna4VtsygxDXB0W5j6kFvWN0uERD2mXeb4IjDuDfvL67GIcHRqmw3/50ANiKSHAAAAAAAYOA++ugjnTp16nPHc7lcy/JrEyKD/J471GjfLtmqLlc1eXNShmHILtoyJhqbjTtl5/9n7+5j5DgPO8//qvplRuwhOeSQYk+PPCPJEiWb5oYckYzWDkOuZUd32NXiQo6wkohbBRQSIIj3hOztLnDJKYoAJUAOB+S0SbxebyAiCEHFYmhgsbewsjxZUaDV2rImRGCBThwzFhOqe/g+w+6urq6qrro/eno8ak2/Vb/PfD9AgT1d9VQ99VSNDD+/eZ5HxTtFOXlHrvXxsMMwDaUOrn0PndBsp3ernaX1Opq70dE7SB3ttQxy3aqFac9eP/NhMwwBRy2thGPAekTIAQAAAAAANqzJ2Um5lit70ZZ1w1J0JCrf8xUfi8uMmgr8QJ7tyck7Ki4V5eSWgw7Lk2eXt+S+9tbjaNegd1APev3qGcS6D2Kdht0wBxwACDkAAAAAAMAGt+fJPbp0/lI52IiUFx6Pj8VlxkwpkEpuSa7lysmVg47KqA57ydbep/b2te6tdpb2eiqrMPWr9xfpg9I5HLae7U7X1Knpjdo5Xzvq3Xu7bRr2nSHgAIYfC48DAAAAAIANbe71ObmWq2wmq9xCTtl0VtmPssqmyz/nr+ZlXbdUuFlQ4VZBxaWi7CVb8qVjZ451tW7DPMVQu/WrvvdureVQ67xhF/Vutp5rHVfvmoP+vCsatafUuzZtVG5YAo5m7wPYqBjJAQAAAAAANryT75zU6cdPK3slK9dyFUvEFI1HJVMKSoFKbkme5a2M4AiCQM+9+1zHrt+ok7K6U3T18Y3+Or4f2qnf6tEOa5Vt9Bf5jb6vFyC0Imw9G5VbS7+ed9g2Ddvp3q02bfTMW23TMO3S7jNspk0HPQADuoWRHAAAAAAAYMNLHUzpxBsnlLuR0+KHi+XRHOmscpncymiO/PW8spms3JyrZ88/q+Rs99fiuPz8xuy4rNeZ3ctrNbpe2Hq2c81BV6/+/WrTYdfMva+H+wTCYiQHAAAAAACApKlDU3qh+ILOPX1OH3zjA0VHowr8QJJkmIY829Pep/Y2PUVVM52OYTsme9nZ369ztXqOdq7Zj7KdWlejHWGDg3bP263yvWrTMGX71SbARkDIAQAAAAAAsMrx147r+GvHtXBhQen305LKIz2S+7o/cgMAALSGkAMAAAAAAGANyf1JJfcTbAAAMMgIOQAAAAAAGDCGYax8DoKg5ePqlV+9r9H+etfGxtLKQtJMqwMA6CUWHgcAAAAAYIBUQoZKwFAdSjRzXBAEdQOKyv61Ao7V+2pdGwAAYFAwkgMAAAAAgAFTCR8aBQ3NHtfqdVuRmc8oPb+8bsWBlCZnJ9uuBwYPozMAAIOKkAMAAAAAgA2mE1NSnZ07q4vnLiq2KSYZkpZP41qu9jy5R3Ovz3WgpgAAAPURcgAAAAAAsIHUmqKqWq3vr7x7RaeOnFJiR0LTh6cVH4vLMA35ri8n58hetHXp/CW9PPKyTr5zUqmDqa7dCwAAACEHAAAAAAD4mHoBx5knzmhydlITuyeU2JVQbFNMgR/IzbkqLBZk3bAUH4srm8nq9OOndeKNE324AwAAsFEQcgAAAAAAMGAqIUOjdTaaPS7Mtddy6sgpTc5OanJ2Ujse3qHNU5tlxky5eVeFWwWNXB9RdCQqM2JKkrJXsnr18KvSr3WsegAAAB9DyAEAAAAAwACphBaV4GJ14LA6gGh0XPXnyv7qQGStcmut2XF27qwSOxOa2D2hHQ/vUHI2qfGZcZWckvLX8oqORmUYhnzPV8kpybM9uZYr+44t56wjsUQHAADoAkIOAAAAAAAGTK2RFNXfN3tcJ/ZdPHdR04enldiV0OapzRqfGdfW6a2yF22VnJJcy5WTdRQfi5e3RFyxREyj20blXHRqnhcAAKAdZr8rAAAAAAAABltmPqPYppjiY3HFNsVkxkyVnJLsRVue7Snwl0eVRAyZUVNmzJQZNxWNRxWNR6WoZCx0bkotAACACkIOAAAAAAAGxEMPPaRMJrOyVSwsLOjtt99e+bnXn9PzacmQDNMoLzKed5W/llc2nZV1w5KTc+TZnnzPV+AHUrA85ZWpcvBhmFI6VJMAAADUxXRVAAAAAAAMgBdffFHpdLqpKaj68jmQfNeXmysvMh4djcq1XBmGIc/2VLxTlJt35dmeSm6pHHiUgnLoAQAA0CWEHAAAAAAADIhUKrXm98lkUslkcuXno0eP9vRzZkt5VImTc1RYLGjk+ogMw5CTdWREyouNu3lXxTtFOXlHrlUVdgSBghRhBwAA6DxCDgAAAAAAUNfk7KRcy5W9aMu6YSk6EpXv+YqPxWVGTQV+IM/25OQdFZeKcnLLQYflldfs8AIp2fg6AAAArSLkAAAAAAAADe15co8unb9UDjYi5YXH42NxmTFTCqSSW5JruXJy5aCjMqrDXrKlPf2uPQAAWK8IOQAAAAAAQENzr8/p5ZGXlc1kJUme7SmeiMuMmzKM8pRVnu3JtcrTVhWXiuWAw5c0J4nZqgAAQBeY/a4AAAAAAAAYDiffOSk35yp7JatsOqs76TvKpXPKZrLKLeSUv56Xdd1S4VZB1i1LQRDouXef63e1AQDAOkbIAQAAAAAAmpI6mNKJN04odyOnxQ8XlU2Xw45cJlcOOa7mlb+eVzaTlZtz9ez5Z5WcZTEOAADQPUxXBQAAAAAAmjZ1aEovFF/QuafP6YNvfKDoaFSBX56LyjANebanvU/t1bEzx/pcUwAAsBEQcgAAAAAAgJYdf+24jr92XAsXFpR+Py2pPNIjuY+RGwAAoHcIOQAAAAAAQGjJ/Ukl9xNsAACA/iDkAAAAAAAAPWMYxsrnIAhCHVdr3+rvq/fX2wcAAIYXIQcAAAAAAOiJStAQBIEMw5BhGGsGDfWOqy6z+udmw5DqfQAAYHgRcgAAAAAAgJ5ZHUjUCxpqHdfs6ItaAUqjfatl5jNKzy+vN3IgpcnZyTWPm3mlqSp9wuXnG5e9/Hxz51rrPI3KhinTilr31s17Wqtcr6/XrWu22571ztfquxLm3Wq3fDeeRaff0W63S61zdPL3FhhGhBwAAAAAAACrnJ07q4vnLiq2KSYZkpbzENdytefJPZp7fa5ndZl5JVwnbaOytco1ul6zGtVLCndfYe6pkbBtWK9sp59bu+3ZyvXDHNdJnW7TWuXaadNBapfKPoIObGSEHAAAAAAAYCjVm+4qzCiOK+9e0akjp5TYkdD04WnFx+IyTEO+68vJObIXbV06f0kvj7ysk++cVOpgSlLjjtBmOh/rdcI204HZSidurb/gb+V6zWjULs10PoepX2Vfqx3RtYKFbtVzrWtWn7fRsa1eL6xOtWkjYds0bLl227RX7VKvbC+ePzDozH5XAAAAAAAAoFXNTjnVrCvvXtGZJ85ocnZS933pPk0dmlLqQErJfUnt/MxObX9wu8bvG9fE7gkldiV0+vHT+ui9jzp2/bU0O/VNreNa7WDvZAdpMx3DrZ6jmfsJ26neSgd4o+O68dw60Z7NXr/63P3oOA/7brZSrp027XW7dOI9BdYzQg4AAAAAANAzlfU1Gi38Xe+4TgccknTqyCltf2C7JmcnlTqQ0j2P3qPJRya187M7te2BbRqfGdeWqS0aS45p8+Rmxe6K6dXDr3a0Dr3QbAd7P6bjWX3dQatf9fUGtZ7NGsSO8bBtOuzPopM2wj0Ca2G6KgAAAAAA0BOVRcQrwcXqoGJ1cNHouNX/1jtPtVr7zs6dVWJnQhO7J7Tj4R1KziY1PjOuklNS/lpe0dGoDMOQ7/kqOSV5tifXcmXfsfXNp76pY39yLGyThBZmHQb0X7+fW7/DId7XMtoF6CxCDgAAAAAA0DO1Aojq75s9rpX9tfZdPHdR04enldiV0OapzRqfGdfW6a2yF22VnJJcy5WTdRQfi5e3RFyxREyj20b1/de/37WQo5UO6WHqNOWvzX+iE8+t1UXE+/luDEIdmtGvMGjQ2wUYVIQcAAAAAABgw8rMZxTbFFN8LK7YppjMmKmSU5K9aMuzPQX+8qiSiCEzasqMmTLjpqLxaHkbjWrhwoKS+5Nt1aNep0vY5moAACAASURBVGq9js9Gi4sPcqfpINTt8vPltlpr0eZudnR347mFXeC+W3rZnt3SjfYK0y79ek+BYcGaHAAAAACwjlWm/Glm/YO1jqtXfvW+etdodG1sDA899JAymczKVrH6u9Xf3759W2+//fbKz936nJ5PS4ZkmIYCP5Cbd5W/llc2nZV1w5KTc+TZnnzPV+AHUrD8TpuSESmXSb+fbqdpOqKyEHK/Fopu1iCHL5VO5MrWC+0+t2bbs9cd4Y0WyB7kjvluvqOdaJd+vKfAoCPkAAAAAIB1qno9g0YhxFrHBUFQd9qg1Vu9c2Nje/HFF3XkyJE135d671H1cV37HEi+68vNuSrcKii3kFM2nVX+al6FmwUV7xTl5l15tqeSWyoHHqWgHHp0SHVn9+pO70YdmfU6TgepE3RQA45aAUO3A6N2n1urAccgtPsg1KGefrVVs6Nw+vGeAsOA6aoAAAAAYB2rXsi53eNqWWtB58p3BB2QpFQq1dL327Zt09GjR1d+7tbnzJby6BEn56iwWNDI9REZhiEn68iIlBcbd/OuineKcvKOXOvjYYdhGkodXPseOqHeNDXDZJA62msZ5LpVC9Oe9YKTYXg+3TYsbTDo9QP6gZADAAAAANBxa4Uetdy+dFvf+4Pv6cdv/Vi3Lt2SJG1/YLvu+yf36dC/OqTxe8e7WVVscJOzk3ItV/aiLeuGpehIVL7nKz4Wlxk1FfiBPNuTk3dUXCrKyS0HHZYnzy5vyX3trcex3g1L5/FaBrHug1inYTfsbTrs9QfaxXRVAAAAAIC2VAcarQQc5//deX394Nd14dQF2Yu2tt27TVs/tVXWVUvzX5/Xf9z/H/Xmr73ZraoDkqQ9T+4pr8Fx3VqZqiqbziqbySq3kFP+el7WDUuF2wUVl4orozrsJVt7n9rblzqH7cxsNB1SpztLWz1fr+sXVth6tlvvMPdfayq01efo1ZRH9UaTtNumYd+ZQXinBmlaOWAYEXIAAAAAADquejHytaas+uMv/7Eu/OEFJfcl9cD/9IDuf+x+TR2aUnJ/UslHkkr+VFJju8b03d/7rs78z2d6fQvYQOZen5NruSuhRjadVfajctCRW8gpfzUv67qlws2CCrfKQYe9ZEu+dOzMsa7WrZW1Gdb6rl7HbXW5Tne0ttt53O36NTpv2EW9m61nq89tEDrjm9GoPaXetWmjcr1s03bbZdCDP6CfmK4KAAAAANaxZtfF6OT6GbXW5ljt9OOntfBXC5o5OqOJByc0Njmm6GhUJaek4lJR1k1L1mZLkdGIjKihy+9c1pl/dkY62Hb1gDWdfOekTj9+WtkrWbmWq1gipmg8KplSUApUckvyLG9lBEcQBHru3ec6dv2wi4tXyrUSADQq14nO0tXnbeav96u/C1O/ZjqRa5UPE6CErWeY59ZOe7YjbJuGDaS61aaNnnmrbdrrdulUeWC9YiQHAAAAAKxTlWChElxUTynV7HGrR2NUhyCtTE1V8YNzP9CV71zR1KEp3f25u5U6kNI9P32PkvuSmtg9oa0zW7VlaosSuxJK7Exo08QmjU2O6cM//1D6m5YuBTQtdTClE2+cUO5GTosfLq5MWZXL5FZGc+Sv55XNZOXmXD17/lklZ7u/FkejaYTqdfqGLTcIelm/sG3YqGy3rjnoGr13/WjTQdDNdhmUewT6hZEcAAAAALCO1Qogqr9v9rhW9691zFu/8VZ57Y3prdr+6e3a+bmdmtg9ocLNgrLprAI/UMkpyS24ci1XTt6Rk3NUTBTlvulKuxteEghl6tCUXii+oHNPn9MH3/hA0dGoAn85BDQNebanvU/tbXqKqmY6HjvROdnu+hzd0I/7auea/Sjb6roa3RC2Y73d83arfK/adNjaBVjvCDkAAAAAAD2Ty+R069ItzRye0V3b7tLI+IjiibgkyYyaioxEFB2NfnK7K6pYIib9vWTk259SC6jn+GvHdfy141q4sKD0+2lJ5ZEeyX3dH7kBAABaQ8gBAAAAAOiZ9Hx6JcwwIoZ8z5e9aEuG5Hu+PNuT7/lSsDw9lmnIjJgyo+XNiBgK0q1NjwWEldyfVHI/wQYAAIOMNTkAAAAAYJ05evSonnjiCc3Pz69sFfPz87pw4YJeeumlle96+fm1P3hNZsQsT0lVLC8ynlvIKZvOyrpmqbhYlJt35dmeSm5JQSkoTxe0HHpEjIh0K1SzAGjDzCvNbwAA9BIjOQAAAABgnTly5EjNfY888ogkaf/+/Svfvfjiiz37/PSvPK0//Ys/lVfwVLxTlHXDkhkx5eQcmRFTJackJ1deg8O1fhJ2+J4v3/NV8kvS9sZtAAAAgI2BkAMAAAAA0DOpR1LyS76Kd4oq3CootimmwA8Uz8ZlRk0FpUBuwZWTdcpb3pFneeWwwykp8AMFKaarAnqNBY8BAIOKkAMAAAAA0DNjk2OaeGBChRsF5bfmZUZNldyS4mNxRWKR8jRWTklO3pFzZ3lbHtXhFlxpQlKi33cBAACAQUHIAQAAAADoqS/+9hd17plzim2OyTAMeUVvJeSQJN/15dqunJyj4p2i7Du2nJwjr+BJP9/nygMAAGCgEHIAAAAAAHrqoX/+kO49cq8u/8VlBX55eqp4Iq5IPCIZUlAqj+ZwreWgY6ko66alT3/50/qbB/9GYrYqAAAALDP7XQEAAAAAwMbzzH99RtM/M62lv19SLp1TNp1VNp1VLpNT7mpO+at55a/nZd2wlL+R16cf+7Se+s9P9bvaAAAAGDCM5AAAAEPNMIyVz0FQ+097ax3XTPl2ygIAajvxrRN689ff1PtffV+5qzlFRiKKjpT/b6pneyoVS4rcFdEX/u0XdPSlo/2tLAAAAAYSIQcAABhalZAhCAIZhiHDMNYMG+odV/l3dWBRXbbWOasDD4IOAGjdY7/1mA780gG999X39OGbH+rm396UJG3fvV33P3a/Dn3lkLbcs6XPtQQAAMCgIuQAAABDbXVYUSuoaOW41eoFF60GGpn5jC6/fVmX//tlSdLM4RnNHJ7R5COTLZ0HANajrTNb9eXf+XK/qwEAAIAhRMgBAABQR7tTUl2/eF3f+F++oWwmK0mK3RVT4Ae69MYlyZS2pLbo6f/ytCZ2T3SszgAAAAAAbBSEHAAAAHU0MyVVre/nvzavP/vXf6at927Vp77wKcXH4jIMQyWnJCfrqLBYUH4hr6/91Nf0T3//n2rfc/u6ei8AAAAAAKw3hBwAAABtqBVwvPd77+nbv/Ft3fOP79H2B7ZrLDmm6F1R+Z4vJ+eocKugkRsjim2KKXY1pm/96rfkeV4f7gAAAAAAgOFFyAEAAIZaJWRotM5Gs8eFuXa1/PW8/tu/+W/61Oc/pV3/aJd2fnanNk9tlhk15WQdWTctxRNxReKR8jn8QL7n61tf+Zb0v0sa7VgVAQAAAABY1wg5AADA0KqEFpXgotbUUo2Oq/5ca5Hytcqttf9P/8WfavM9mzV+37gmHppQcl9S4/eNy7Vc5RZyMmOmAj9QyS3Jsz25BVeu5Sp+Oy77rC39rx1oHAAAAAAANgCz3xUAAABoRxAEK1v1980eV721U84reLr8F5c1Pj2uxN0JJe5OaCw5pk07Nmlky4jim+OKJWLlbdPHt5GtI9KPJZU61DgAAAAAAKxzjOQAAADooPT7aUVHo4ptiik6EpVhGHLyjvLX8yoVS/JdXwrKI0DMiCkzaioSi5S3eERG1JDS/b4LAAAAAACGAyM5AADA0MlkMvrggw/0ox/9aGWr+NGPfqTvfe97Kz+/9tprPf189a+uyjRNyZBKbklOzlH+Wl7ZdFbWDUvFO0V5BU8lpyS/5Eu+pECSIRmmUS67EKJRAAAAAADYgBjJAQAAhs7Xv/513X333XrmmWc+sW/nzp3atGnTys+PPPJITz8Xv1eUX/JVKpYDDuumpUg8IifvyDAMlYolFbNFuXlXXsGT53gqeSUFpUCBvzztFQuPAwAAAADQFEIOAAAwlK5evaqtW7d+4vutW7d+7Pvdu3f39PMN/4aCIFAxW1ThVkHxsbgkKZ6Ny4ya8j1fruWqeKcoJ+fItVx5tievWB7dEfiBgtTH1/8AAAAAAABrI+QAAADooIkHJxSJRmTftlW4UVB0JCrf8xUfiysSi/xklEfeKQcdWUdubnlUh+0piATS9n7fBQAAAAAAw4GQAwAAoIOMiKHZX5rVX379LxUfi8swDXlFbyXkkKSSU5JX8FTMFVVcKqqYLYcdTtaRDkoy+nsPAID+M4yf/I9BENQe4VfvuG7sAwAAGDSEHAAAAB32c//3z+n7Z76vO/9wR0EQyLM9xRIxReIRyZACL5BX9MrTVmXLQYd929ZdE3fJ/ZJbXogcALBhVUKGIAhkGIYMw1gzbKh3XHWZTuwDAAAYRIQcAAAAXfArF39FX/3MV7X04ZK8u38SchimocAPVkZzOHlHhVsFjWwe0S9/8Mv6nf/nd/pddQDAAKgEC5UAo9Xj6gUTYffVkpnPKD2fliSlDqQ0OTu55nEzr7R8aknS5ecbl738fHPnWus8jcpWl2n2Ws2qdW/dvKe1yvX6et26ZrvtWe98nX5X2n2ve/UsOv2OtvIs+vG+ARg+hBwAAABdMDo+qq/87Vf09Ue+rmw6q0g8olgiJiNiSIHku76cvCPf87V1eqt+8Xu/qNimWL+rDQBA087OndXFcxfL//tlaGUkomu52vPkHs29Ptezusy8Eq6Ttl7ZsKFMKxrVSwp3X/XaI+x9hWnDRmU7/dzabc9Wrh/muE7qdJuG+T1o1KbttEs/3jcAw4uQAwAAoEviY3F95W++ou/87nf0nVe+o6XLS4qPxSVJTs7R+L3jevRXH9VP/28/3eeaAgDWs3pTToXZd+XdKzp15JQSOxKaPjy9sgaV7/pyco7sRVuXzl/SyyMv6+Q7J5U6mJLUuCO0mc7Hep2wzXRghgkzKmW61ZHdqF2a6Xxevb/Z9gh7X7WChW7Vc61rVp+30bGtXi+sTrVpI2HbNGy5dtu0nd+hsMFLddlePH8A/UPIAQAA0GWP/uqjevRXH5UkZf4yI0k1p/IAAKCTuhFwnHnijCZnJzWxe0KJXQnFNsUU+IHcnKvCYkHWDUvxsbiymaxOP35aJ944oalDUx29r9UaTWfVKERp9ftOatQx3EyncPU5GpULc1/12rBb9Qzz3DrRnmtdv1PTpXVaq20aplw7bdruFGHt/G6GbRsAw8nsdwUAAAA2ksnZSQIOAEBDlfU16q3H0ei4TgccknTqyCltf2C7JmcnlTqQ0j2P3qPJRya187M7te2BbRqfGdeWqS0aS45p8+Rmxe6K6dXDr9a9B7Su2U7gXnfqVl9vUOvZrEH8i/+wbTrsz6IZG+EeAayNkRwAAAAAAAyQyiLileBideCwOoBodNzqf1fvD7vv7NxZJXYmNLF7Qjse3qHkbFLjM+MqOSXlr+UVHY3KMAz5nq+SU5Jne3ItV/YdW9986ps69ifHOtE8LQmzDgP6r9/Prd/h0EZ+X/v97AEMJ0IOAAAAAAAGTK2RFNXfN3tcJ/ZdPHdR04enldiV0OapzRqfGdfW6a2yF22VnJJcy5WTdRQfi5e3RFyxREyj20b1/de/37WQI8wURBWD3JHKX5v/RCeeW6uLiPfz3RiEOjSjF+/oMP3OAugfQg4AAAAAAFBXZj6j2KaY4mNxxTbFZMZMlZyS7EVbnu0p8JdHlUQMmVFTZsyUGTcVjUfL22hUCxcWlNyfbKseYef/r1VuGDqTB6FulfUM1lq0uZsd3d14bmEXuO+WXrZnt3SjvYb5dxZA77EmBwAAAAAAAyKTyXxsq/X9d7/73ZV9b7/9dtc/p+fTkiEZplFeZDzvKn8tr2w6K+uGJSfnyLM9+Z6vwA+kYHnKK1MyIuUy6ffT7TRNR1x+/uPboBrkjtxK2FHZeqHd59Zse/Y6YGi0ePogBx69ekeH5XcWQH8RcgAAAAAAMABeeuklvfXWWwqCYGWrWP3dWvt68jmQfNeXm3NVuFVQbiGnbDqr/NW8CjcLKt4pys278mxPJbdUDjxKQTn06JDqDs/VHZ+NOoTrdSgPUmfyoAYctTqZu9353O5zazXgGIR2H4Q61NPLgKPWd4P0Owug/5iuCgAAAACAAfHDH/5QzzzzzCe+T6VSNX8+evRo1z9ntpRHlTg5R4XFgkauj8gwDDlZR0akvNi4m3dVvFOUk3fkWh8POwzTUOrgx++hk+pNpzRMBqmjvZZBrlu1MO1Zr/N8GJ5Pt9EGAAYRIQcAAAAAAKhrcnZSruXKXrRl3bAUHYnK93zFx+Iyo6YCP5Bne3LyjopLRTm55aDD8uTZ5S25r731ONa7Ye48HsS6D2Kdhh1tCmBQMV0VAAAAAABoaM+Te8prcFy3Vqaqyqazymayyi3klL+el3XDUuF2QcWl4sqoDnvJ1t6n9valzsPSGdtq53GjKXsGpTM6bD3brXeY+681Fdrqc/RqXYh6o0nabdOw70w/p6hqteyg/14A6DxCDgAAAAAA0NDc63NyLXcl1Mims8p+VA46cgs55a/mZV23VLhZUOFWOeiwl2zJl46dOdbVurWyNsNa3/Wz07PdOlTfV7fWKmi34zhsPVt9boPwTJvRqD2l3rVpo3L9atN2fmd79XsBYDAwXRUAAAAAAGjKyXdO6vTjp5W9kpVruYolYorGo5IpBaVAJbckz/JWRnAEQaDn3n2uY9cPu7h4pVwrHZ3NdELXumaYazTz1/vV39W7r0Z/kd/o+3oBQivC1jPMc2unPdsRtk3Ddrx3q00bPfNW2zRsu4T9nW2m7KCHXwDCYSQHAAAAAABoSupgSifeOKHcjZwWP1xcmbIql8mtjObIX88rm8nKzbl69vyzSs52fy2ORtMI1ev0HeZOz3r31ctrNbpe2Hqu1+cmNX5f+9Gmg6Qf7xuA4cVIDgAAAAAA0LSpQ1N6ofiCzj19Th984wNFR6MK/ECSZJiGPNvT3qf2Nj1FVTMdj53onGz1HL3oEB22++pH2VbX1eiGsJ3q7Z63W+V71ab9ur9OXBvAcCHkAAAAAAAALTv+2nEdf+24Fi4sKP1+WlJ5pEdyX/dHbgAAAFQQcgAAAAAAgNCS+5NK7ifYAAAA/UHIAQAAAAAA0AGtLJLMdDoAAHQGC48DAAAAAAAAAIChxEgOAAAAAACADmB0BgAAvcdIDgAAAAAAAAAAMJQIOQAAAAAAAAAAwFAi5AAAAAAAAAAAAEOJkAMAAAAAAAAAAAwlQg4AAAAAAAAAADCUCDkAAAAAAAAAAMBQIuQAAAAAAAAAAABDKdrvCgAAAAAAEIZhGCufgyAIdVytfa2cu95+AAAAdBcjOQAAAAAAQ6cSQlQChtWhRLPHVQKKylYdbDQKL2pdEwAAAL1DyAEAAAAAGEqVEKJRGFHruHZGYDCCAwAAYDAwXRUAAAAAAC1oJeC4fem2vvcH39OP3/qxbl26JUna/sB23fdP7tOhf3VI4/eOd7OqAAAA6x4hBwAAAABgw2s2uGgl4Dj/787rL//wL6VAGh0f1bZ7t8kv+bKuWpr/+rwuvHpBB375gB777cfarT4AAMCGRcgBAAAAANjQWp16qnotjrXK//GX/1iZ+YyS+5Ia2zWmeCIuSXJtV8U7Rdm3bFk3LH33976rqxeu6plvPdP+jQAAAGxAhBwAAAAAgKFUCRcaLQBe77hWA47qY9cqf/rx01r4qwXNHJ3RxIMTGpscU3Q0qpJTUnGpKOumJWuzpchoREbU0OV3LuvMPzsjHWy6GgAAAFhGyAEAAAAAGDqV0KISXKwOGlYHD42OW/3v6v2rv1urbC0/OPcDXfnOFU0fntbdn7tbu/bu0pZ7tkiGZC/ayl/LKzoalWEaCvxAvuur5JT04Z9/KG2RtDtEYwAAAGxghBwAAAAAgKFUK3So/r7Z45rdV++4t37jLW27d5u2Tm/V9k9v187P7dTE7gkVbhaUTWcV+IFKTkluwZVruXLyjpyco2KiKPdNl5ADAACgRWa/KwAAAAAAwHqQy+R069Itbbp7k+7adpdGxkdW1uIwo6YiIxFFR6Of3O6KKpaISTclI19/6i0AAAB8HCM5AAAAAADogPR8eiXMMCKGfM+XvWhLhuR7vjzbk+/5UlCeAsswDZkRU2a0vBkRQ0G6+fVBAAAAwEgOAAAAAMAQOXr0qJ544gnNz8+vbBWrv/vBD36gl156aWVfLz4v/t2izIhZnpKqWF5kPLeQUzadlXXNUnGxKDfvyrM9ldySglKgwA9WQo+IEZFutdM6w6GyRkozC8bXOi7MvtXfN7o2AAAYHozkAAAAAAAMjSNHjtTc98gjj3zs5xdffLGnn3/4//6wPGKj4Kl4pyjrhiUzYsrJOTIjpkpOSU6uvAaHa/0k7PA9X77nq+SXpO0Nm2CorV7EvRI2rLX+Sb3jqss0u69yPgAAsL4QcgAAAAAA0AGpR1LyS76Kd4oq3CootimmwA8Uz8ZlRk0FpUBuwZWTdcpb3pFneeWwwykp8AMFqfXfCV8JGioBRqvH9TKoyMxnlJ5PS5JSB1KanJ1c87iZV8Kd//Lzjctefr72vlpl65Vpp1y9c7RSttVzt3qNtco3UzbsPXXqet28Ziee+VrnavVdC/tutlO+G8+i0+9ou+1S7zydfPbAoCLkAAAAAACgA8YmxzTxwIQKNwrKb83LjJoquSXFx+KKxCLlaayckpy8I+fO8rY8qsMtuNKEpES/72I41Ao9GgUizYYlZ+fO6uK5i4ptikmGpOVDXcvVnif3aO71uTbvoHkzr7TWcbl6XyfLNVO+E7pVv1rt2Oia9TSqa6frGfaa7bZps+cKc1wndbpNu9GevW6XTj57YJARcgAAAAAA0CFf/O0v6twz5xTbHJNhGPKK3krIIUm+68u1XTk5R8U7Rdl3bDk5R17Bk36+z5UfImGmq1or8Kj+7sq7V3TqyCkldiQ0fXha8bG4DNOQ7/pyco7sRVuXzl/SyyMv6+Q7J5U6mJLUuEOzmU7Eep2p9Tppw5RrVN9GncKrz9GNTtuw9as1wqDZ+wp7T9Xn7HY917pm9XmbOb7Va4bRqTZtJGybhi3Xbnu22i7NhDSt/DdidbluPXugl1h4HAAAAACADnnonz+ke4/cq6XLS8pmssqms8p+tPxvJqvc1Zys65YKNwuyb9sqLhZl3bR0/5fulx7sd+03tivvXtGZJ85ocnZS933pPk0dmlLqQErJfUnt/MxObX9wu8bvG9fE7gkldiV0+vHT+ui9j7pap0adpK12arazr5nrdkK79VvruGYCg7Cd6q10gDc6rtkpi1p97p1o02auX33efnSct9qmYcq10569bpdOPXtg0BFyAAAAAADQQc/812c0/TPTWvr7JeXSuXLAkc4ql8kpdzWn/NW88tfzsm5Yyt/I69OPfVpP/een+l3tnqlMGVVvPY56xzUqF9apI6e0/YHtmpydVOpASvc8eo8mH5nUzs/u1LYHtml8ZlxbprZoLDmmzZObFbsrplcPv9qVuqB1zQYA/Z4uaFDr2axB7BgP26bD/iykwa4b0EtMVwUAAAAAQIed+NYJvfnrb+r9r76v3NWcIiMRRUfK/xfcsz2ViiVF7oroC//2Czr60tH+VraHKmtmVIKKelNL1Tqu3rob9fZVhyOr952dO6vEzoQmdk9ox8M7lJxNanxmXCWnpPy1vKKjURmGId/zVXJK8mxPruXKvmPrm099U8f+5Fhb7QI00u/1E/odDg1iuDJIaB9sdIQcAAAAAAB0wWO/9ZgO/NIBvffV9/Thmx/q5t/elCRt371d9z92vw595ZC23LOlz7XsvVoLfld/X29h8DD76pW5eO6ipg9PK7Eroc1TmzU+M66t01tlL9oqOSW5lisn6yg+Fi9vibhiiZhGt43q+69/v2shR7sLYrezrsEgGvT69VKnQoBm2rTfAcug1KEZw/SODlNdgUYIOQAAAAAA6JKtM1v15d/5cr+rgToy8xnFNsUUH4srtikmM2aq5JRkL9rybE+BvzyqJGLIjJoyY6bMuKloPFreRqNauLCg5P5kW/UIs2h0vbLtdAYPekfyINTv8vPldl9r0eZudh7XOne7IUCYhc67pZft2S29aK9OBT+D8PsEtIs1OQAAAAAAQNdlMpmPbWG/f/vttzv6OT2flgzJMA0FfiA37yp/La9sOivrhiUn58izPfmer8APpGB56itTMiLlMun30+02T8e12jE86H8pP8j1q4Qdla0XKgtYt7OQdTNt2uuAodFC7oMceAzyO1ptmOoKNIORHAAAAAAAoKteeuklPfjggzp69Ogn9rU6vVT1Ghyd+KxA8l1fbs5V4VZB0dGoXMuVYRjybE/FO0W5ebe8nopbKgcepaAcenRIrc7GWqMFGpVtptzqY+vVod8GtX61Fqbu9oLVtYKATj/zQWr3yv0NqkFqq0aGqa5Aswg5AAAAAABA1/3whz/UM88884nvU6nUmsfX+n51UNKJz6kD5es4OUeFxYJGro/IMAw5WUdGpLzYuJt3VbxTlJN35FofDzsM01Dq4Np17YRWO69bLTfoHZ6DXj9psOu2llbbtF64MAzPp9v60QbtrruzkZ8X1idCDgAAAAAAsGFNzk7KtVzZi7asG5aiI1H5nq/4WFxm1FTgB/JsT07eUXGpKCe3HHRYnjy7vCX3tbceR78MeofnoNevnkGt+6DWa1gNU3sOU12BVrEmBwAAAAAA2ND2PLmnvAbHdUu5hZyy6Wx5y2SVW8gpfz0v64alwu2CikvFlVEd9pKtvU/t7Xf1Qxn0Ds9W69doiqhBud+w9exEvcO0aa2t+phuqzeapN02DfvO9OudCjNt16C8/0C3EHIAAAAAAIANbe71ObmWuxJqZNNZZT8qBx25hZzyV/Oyrlsq3CyocKscdNhLtuRLmCb6XQAAIABJREFUx84c62rdGnXAtlpu9b5B7fBst37V996ttRzaDVTC1nOt4wa1Q74VzbzrvWrTZn+/+tmerU43NsjPHmgX01UBAAAAAIAN7+Q7J3X68dPKXsnKtVzFEjFF41HJlIJSoJJbkmd5KyM4giDQc+8+17HrN+pUrbW4eCPV5VaXaeav45u9Zq3Ft1vVTv1WL069Vtlu3FOYACVsPRuVq6XdZx5G2DYNG0h1q00bPfNW27Pbvz/1zturZw/0AyM5AAAAAADAhpc6mNKJN04odyOnxQ8XV6asymVyK6M58tfzymaycnOunj3/rJKz3V+Lo9Z0QI06JXs1jdCgqdeZ3ctrNfN8Wvm+E9ccdPXq3682HTSMygDWxkgOAAAAAAAASVOHpvRC8QWde/qcPvjGB4qORhX4gSTJMA15tqe9T+1teoqqZjoi2+msDFO23c7RbneuduL8rZ6j18+g3bK9vL+w5+zVvfXjuv16X8KUJwzBRkHIAQAAAAAAsMrx147r+GvHtXBhQen305LKIz2S+7o/cgMAALSGkAMAAAAAAGANyf1JJfcTbAAAMMgIOQAAAAAAGDCGYax8DoKg5eNaKR+mHNCsVhaSZmodAEAYLDwOAAAAAMAAqQQNlZBhdfDQ7HFBEDQMKdY6bzPlAAAABgkjOQAAAAAAGDCVoCEIgpohRyvHVauM4GilTC2Z+YzS88vrVhxIaXJ2su1zYv1gdAYAoNsIOQAAAAAA2ECqp6gK6+zcWV08d1GxTTHJkLR8StdytefJPZp7fa7tawAAADRCyAEAAAAAwAbRiYDjyrtXdOrIKSV2JDR9eFrxsbgM05Dv+nJyjuxFW5fOX9LLIy/r5DsnlTqY6lDtAQAAPomQAwAAAOigbi8WXG8/CwYDaEb1FFWtBB9X3r2iM0+c0eTspCZ2TyixK6HYppgCP5Cbc1VYLMi6YSk+Flc2k9Xpx0/rxBsnunEbAAAAkgg5AAAAgI5ZvQiwYRg1Ow7rHVdvoeHq863+ud4+AMOn2TUzWl1bY61wtJX/Vpw6ckqTs5OanJ3Ujod3aPPUZpkxU27eVeFWQSPXRxQdicqMmJKk7JWsXj38qvRrTV8CAACgJYQcAAAAQAd1c7HgboQWN394U5I0sXui4+cGEM7qALTyc0V1KFrvuOrPzfw3pF65s3NnldiZ0MTuCe14eIeSs0mNz4yr5JSUv5ZXdDQqwzDke75KTkme7cm1XNl3bDlnHYklOgAAQBcQcgAAAABDpBNTUl34Txf0P373f+jmj24qGi//XwLP8TTxwIQ+/28+r30n93WkrgDCq/X7Xf19s8c1e5165S6eu6jpw9NK7Epo89Rmjc+Ma+v0VtmLtkpOSa7lysk6io/Fy1sirlgiptFto3IuOk3VBwAAoFWEHAAAAMAQafQX3fV4RU9/+NN/qKV/WNJd2+7S1IEpmXFzZbHg/PW8/uxf/5ne+4P39Ivv/aKMSHMjTACsf5n5jGKbYoqPxRXbFJMZM1VySrIXbXm2p8BfHlUSMWRGTZkxU2bcVDQeLYepUclY4L8pAACg88x+VwAAAABAZwRBsLJVK2aL+vf3/XsVF4ua+ZkZ3f/Y/Zp6dErJfUnd/bm7tePhHbr7s3dr671bdfvHt/W7078rJ89fXgO99tBDDymTyaxsFX/913+98vntt9/u+ef0fFoyJMM0youM513lr+WVTWdl3bDk5Bx5tiff8xX4gRQsjzwzVQ4+DFNKt94eAAAAjTCSAwAAAOigbi0WvLpMmOP+w+f+g2KJmO559B5NPDShzanNisQj8mxP9qIt64ZV/uvsuCnDMLT0D0v62t6vSc82XT0AbXrxxReVTqcb/p6v3t/Lzwok3/Xl5sqLjEdHo3ItV4ZhyLM9Fe8U5eZdebanklsqBx6loBx6AAAAdAkhBwAAANAh3V4suDoUqVWuuoP02//nt1W8U9T9X7pfO/fsVPKnktryqS0K/ECFmwXlruVkRs1yB2ZlweCip6XLS9KfSzrSTqsAaEUqlVrz+4cffnjl89GjR3v+OXWgXC8n56iwWNDI9REZhiEn68iIlBcbd/OuineKcvKOXKsq7AgCBSnCDgAA0HmEHAAAAEAHdXux4FbL+Z6v97/2viZ2T6wsFDzx0IS23b9NhVsFGaZRDjUsT26+vGhwMVtUfCyuka0jct9zZfws8+gDG93k7KRcy10Z+RUdicr3fMXH4jKjpgI/kGd7cvKOiktFObnloMPyymt2eIGU7PddAACA9YiQAwAAAFjHbv/dbXmWp9FtoxrZPKLYppgMw5BrueWFgk2jvEBwzFQkHlFkJKLoSFTRkahid8WkG1Jwm7++BiDteXKPLp2/VA42IuWFx+NjcZmx8kiwkluSa7lycuWgozKqw16ypT39rj0AAFivWHgcAAAA6IBsNvuxreLatWsrny9fvtzzz5n5jGRIkVhEkuQVPVk3LWXTWRVuFuTmXZWckgJ/ecHy5YWFjYghM2qyWDCAFXOvz8m1XGUzWeUWcsqms8p+lFU2Xf45fzUv67qlws2CCrcKKi4VywGHL2mu37UHAADrFSEHAAAA0KY/+qM/0oULF5TJZFa2ih/96Ecrn996662ef7ZuWDLM5fnyC67s2/ZK52T+Wl6F2wU5WUdewVPJKcl3ffklX/LLU2CZMmVYTFcFoOzkOyfl5lxlr5TDjTvpO8qlcyvBR/76ctBxqyDrlqUgCPTcu8/1u9oAAGAdY7oqAAAAoE0ffvihfvzjH+s3f/M3P7Hv85///MrnX/iFX+j5513/aJcCPyjPk79YVP56XoZpyMk5K9PNOHlHxTtFufnlhYKdkkpuSYEXqBSUFOxiuioAZamDKZ1444RePfyq7Du2RreNKhqPyogYCvxAvufLs72VERzPvfuckrNJ6b/0u+YAAGC9IuQAAAAA1rHJRyZVckoqLhVl3bQUvSuqwA8Uzy4vFlwqLxZczC7Pn19ZLNj25BU9BaVAxhQjOQD8xNShKb1QfEHnnj6nD77xgaKj5f+uSOXp7jzb096n9urYmWN9rikAANgICDkAAACAdSw+Ften/vGndPNvb2pk84jMqKmSW1I8sWqxYKe8WHAxW1RxqSgn68jJO3JyjjQtBVFGcgD4pOOvHdfx145r4cKC0u+XF+9JHUwpuS/Z55oBAICNhJADAAAAWOd+/vTP6/d3/75y13KSUV58PJ6IKxIvL0ZemV6mMm1VJejwbE863ufKAxh4yf1JJfcTbAAAgP5g4XEAAABgnds6vVU/++LPKpcpLw6cTa/aKosFX8vLulFeLLiwWFAxW9QXf+uL0uZ+1x7AemMYxsoW9rha+1Z/3+j8AABgfWAkBwAAALABHP4/Dis2GtO3f/3bcrKORjaPKDISkWEaCoJAvrs8miPryLVd/dz/9XM6+JWDOv/S+X5XHcA6UgkegiBYCSKC4JNT4tU7rrpM9c9rnQ8AAKxfhBwAAADABvHorz6q6S9M6+y/OKvcQk4lr6RIrDxlVckpf07sSOhf/n//UslZpp4B0B2VEKISYLR6XKdDjMx8Run55TVFDqQ0OTu55nEzr4Q7/+XnG5e9/Hzj86x1jlrlmq1rM9dtpNa1mj13K/dVr1yvr9eta7bbnvXO1+gcrbZpu+91r55Fp9/RVp5FO+8NgOYRcgAAAAAbSOpQSs//3fP6+//+9/rxmz/WP7z7D5KkT33hU7rvi/dp+mem+1xDAGhPs4HI2bmzunjuomKbYpIhaflQ13K158k9mnt9rss1/YmZV8J10jYq1231Orkr+zp9X2HDpkZ17Ub7t3rNdtuzleuHOa6TOt2mtcq106bttEs77xuA1hFyAAAAABuNIU3/zDSBBoCh12iqqrWmw7ry7hWdOnJKiR0JTR+eVnwsLsM05Lu+nJwje9HWpfOX9PLIyzr5zkmlDqYkNe4IbabTsl4nbDOdtKv31yvXTCdxpzpZG7VLJ+9rreu22hFdK1joVj3Xumb1eRsd2+r1wupUmzYStk3Dlmu3TcO2S61r9yNUAtY7Fh4HAAAAAABDp9Z6HvVcefeKzjxxRpOzk7rvS/dp6tCUUgdSSu5Laudndmr7g9s1ft+4JnZPKLErodOPn9ZH733UpTsoa7aDuJUgo1ea6Rhu9RzNBAZhO9Vb6QBvdFyzUzHVOq7VunTyntc6dz/ep7DvdCvl2mnTsO0S5tkDaA8hBwAAAAAA6JnKdFL11uNodFyYgEOSTh05pe0PbNfk7KRSB1K659F7NPnIpHZ+dqe2PbBN4zPj2jK1RWPJMW2e3KzYXTG9evjVlq/TKc12lg7ytERr6fR9dUr19Qa1ns0axM70sG067M8CQHcxXRUAAAAAAOiJyiLileBidVCxOrhodNzqf1fvrw5EVpc7O3dWiZ0JTeye0I6Hdyg5m9T4zLhKTkn5a3lFR6MyDEO+56vklOTZnlzLlX3H1jef+qaO/cmxTjZFXw1i5/d60ukpwcJev1/X28jvV7+fPbBREXIAAAAAAICeqTUCo/r7Zo9rdt/Fcxc1fXhaiV0JbZ7arPGZcW2d3ip70VbJKcm1XDlZR/GxeHlLxBVLxDS6bVTff/37XQs51utfnq/X+wqjEyFAq6N1BmFB+kHv6O/FO0oABPQGIQcAAAAAAFjXMvMZxTbFFB+LK7YpJjNmquSUZC/a8mxPgb88ciRiyIyaMmOmzLipaDxa3kajWriwoOT+ZFv1qNep2u3Oz351PA9Cp+7l58v3v9YC093s6K517naeRdgF7rull+3ZLd1or248ewC1sSYHAAAAAADouoceekiZTGZlq1j9XeX7S5curex/++232/6cnk9LhmSYhgI/kJt3lb+WVzadlXXDkpNz5NmefM9X4AdSsDz1lSkZkXKZ9PvpDrbG+jbIHbmVsKOy9UJlAetuLWRdfVyvNFo8fZADj169o+0+ewDNIeQAAAAAAABd9eKLL+rIkSMKgmBlq1j93eo1OVbv78RnBZLv+nJzrgq3Csot5JRNZ5W/mlfhZkHFO0W5eVee7anklsqBRykohx4dUt3hubrjc5A7hFsxqAFHrU7mbnc+1wsCmnnmrQYcg9Dug1CHenoZcNT6br38vgODgumqAAAAAABA16VSqaa/f+CBB1Y+Hz16tO3PqQPlazg5R4XFgkauj8gwDDlZR0akvNi4m3dVvFOUk3fkWh8POwzTUOrg2vXvhHrTKXVCrzp1B6mjvZZBrlu1MO1Zr/N8GJ5Pt9EGwPpEyAEAAAAAANa1ydlJuZYre9GWdcNSdCQq3/MVH4vLjJoK/ECe7cnJOyouFeXkloMOy5Nnl7fkvvbW41jvhrnzeBDrPoh1Gna0KbB+MV0VAAAAAABY9/Y8uae8Bsd1a2Wqqmw6q2wmq9xCTvnreVk3LBVuF1RcKq6M6rCXbO19am9f6txoaptmOm17MS1Oq53HnbivXghbz3brHeb+a02FtvocvVoXot47126bhn1n+jlFFYDuI+QAAAAAAADr3tzrc3ItdyXUyKazyn5UDjpyCznlr+ZlXbdUuFlQ4VY56LCXbMmXjp051tW6tbI2QytlVutW52u7ncft3lfY61R/3+qi3s3Wc63j6l1zUAKeRhq1p9S7Nm1Url9t2uqzBxAe01UBAAAAAIAN4eQ7J3X68dPKXsnKtVzFEjFF41HJlIJSoJJbkmd5KyM4giDQc+8+17HrN+qMrbVQcaXcWuX72Vm6uj7N/PV+9Xdh7quZzvVa5cMEKGHr2ajcWtppz3aEbdOwgVS32rTRM2+1TcO2S5hnD6A9jOQAAAAAAAAbQupgSifeOKHcjZwWP1xcmbIql8mtjObIX88rm8nKzbl69vyzSs52fy2ORtMI1ev0rWfQ/2o87H11+lqNrhe2nu1cc9A1el/70aaDZD0/e2AQMZIDAAAAAABsGFOHpvRC8QWde/qcPvjGB4qORhX4gSTJMA15tqe9T+1teoqqZjosO9GpGeYcw7D+QKvnaOea/Sjb6roa3RA2OGj3vN0q36s27df9AWgdIQcAAAAAANhwjr92XMdfO66FCwtKv5+WVB7pkdzX/ZEbAACgcwg5AAAAAADAhpXcn1RyP8EGAADDipADAAAAAAAALS2SzFQ8AIBBwcLjAAAAAAAAAABgKDGSAwAAAAAAAIzOAAAMJUZyAAAAAAAAAACAoUTIAQAAAAAAAAAAhhIhBwAAAAAAAAAAGEqEHAAAAAAAAAAAYCgRcgAAAAAAAAAAgKFEyAEAAAAAAAAAAIYSIQcAAAAAAAAAABhKhBwAAAAAAAAAAGAoEXIAAAAAAAAAAIChRMgBAAAAAAAAAACGEiEHAAAAAAAAAAAYSoQcAAAAAAAAAABgKBFyAAAAAAAAAACAoUTIAQAAAAAAAAAAhhIhBwAAAAAAAAAAGEqEHAAAAAAAAAAAYCgRcgAAAAAAAAAAgKFEyAEAAAAAAAAAAIZStN8VAAAAAAAAH2cYxsrnIAhaPm71963sAwAAGDaM5AAAAAAAYIBUQohK+FAdSjR7XBAEK1u1evsAAACGCSM5AAAAAAAYMJXwIQiCmiFHK8e1KzOfUXo+LUlKHUhpcnZyzeNmXgl3/svPNy57+fna+2qVrVemnXKtavc6a5Vvpmx1uV5fr1vX7PRzW32+Vt+ZsO9YO+W78Sw6/Y724ncv7PsNYP0h5AAAAAAAYB2qN+VVs9NhnZ07q4vnLiq2KSYZkpYPdS1Xe57co7nX5zpa53pmXlm7E7NeJ3JlXyfLtard69QqX6s9Gl2znkZ17XQ9w1yz08+t2bYK26bt6HSbdvp3qFHZRucMc72w1wSwvhFyAAAAAACwzqwVaqwe9VFrX8WVd6/o1JFTSuxIaPrwtOJjcRmmId/15eQc2Yu2Lp2/pJdHXtbJd04qdTAlqXFHaDOdz/U6YWt10ja6bqfLtSrsdWqNMGi2fpV9rXYK1woWulXPta5Zfd5Gx7Z6vbA61aaNhG3TsOXabdNW26UTzzDsswCw/rAmBwAAAAAAWHHl3Ss688QZTc5O6r4v3aepQ1NKHUgpuS+pnZ/Zqe0Pbtf4feOa2D2hxK6ETj9+Wh+991FX69RM52qv9rWqE9epPq6Zzt+wneqtdIA3Oq7ZKYtqHddqXTp5z2udux/TIbXapmHKtdOmYdql3WfYr2cBYHARcgAAAAAAMGAq00k1Wmej2eNacerIKW1/YLsmZyeVOpDSPY/eo8lHJrXzszu17YFtGp8Z15apLRpLjmnz5GbF7orp1cOvduz6KPv/2bv34Liuw87zv3v7ARANkCBBio2GTIgSRUmmNZIgUvI6w5CxndXGa6cSkVrL0tQ4RSXeySSz2mxNslvZ1cic0npqd2uSUmbtRHZKdDYcyhJDp8bJlJ1VeS1mFFq2xCiKtcxGHkaiDDXAN4Duvn37PvePRkNgC/1+Avx+qm6p0feee8493XCZ54dzTr0BQLf/ir28vn5tZ736cbC82T5d7Z8FADSL5aoAAAAAAOgjpU3ES8HF8qWkypedqnZd+T3rOXf84HEltiQ0tnNMm2/frORUUqOTo/IdX7kLOUUHozIMQ4EXyHd8ebYn13JlL9j65sPf1IPfeLBNvYC1rp37nrRSf6/q68dwBQBWK0IOAAAAAAD6TKXNwMvfr/e6es+dOXFG2/ZuU2JrQiMTIxqdHNWGbRtkz9nyHV+u5crJOIoPx4tHIq5YIqbBjYP60Qs/6ljI0YmNtDtRrl/rWQ3aEQI0uol4L4OGfmhDPXodBgFAPQg5AAAAAACAZk7PKDYUU3w4rthQTGbMlO/4sudsebanMFicORIxZEZNmTFTZtxUNB4tHoNRzb4+q+Q9yZbaUW2Qs9kB4W6X69d6arVh8umVN3zu5MBzpXu3EgI0u8F9p3SzPzul29/RfvidALB6EHIAAAAAANAnZmZmrvl5fHy87e9PT09rz549kqSTJ09q3759kqSXnntJMiTDNBQGodycq9yFnHzHVxiEcrKOPNtT4AUKg1AKF5e+MiUjUiyTfi3dcsjRLs0OknfrL+z7+S/5ezEI32oQUG9/dvvZKm2eXmrHSqFSv+j2d7SffycA9DdCDgAAAAAA+sDhw4d16623av/+/R841+iyVNXeD4Kg8nWhFLiB3Kyr/JW8ooNRuZYrwzDk2Z4KCwW5OVee7cl3/WLg4YfF0KNNKg1wVpplUOnaavdqd7lG9etgbqWNqTu9YXW1IKCez7zRgKMf+n150NGPCDgArCaEHAAAAAAA9Im33npLjzzyyAfeT6VSK17fzPvLzy0PVPY/sl9v//7bcrKO8nN5DVwckGEYcjKOjEhxs3E356qwUJCTc+Ra14YdhmkotWfletuh3kFvAo7W9XPbyjXTn9XChdXw+XQaAQeA1YaQAwAAAAAAaHxqXK7lyp6zZV2yFB2IKvACxYfjMqOmwiCUZ3tyco4K8wU52cWgw/Lk2cUjeXdvl6oi4Oicfmx7P7ZptSPgALAamb1uAAAAAAAA6A+7HtqlTDoj66Kl7GxWmXSmeMxklJ3NKncxJ+uSpfzVvArzhaVZHfa8rTsfvrOnbV9rAUetJaL6ZXC42Xa22u5mnv/c45WP8ms6rdpsklb7tNnvDAEHgNWKkAMAAAAAAEiSDr5wUK7lLoUamXRGmfeKQUd2Nqvc+Zysi5byl/PKXykGHfa8LQXSg8ce7GjbOrHEUL8GHJXKV/q5XVoNVJpt50rXVatztQyO1+pPqXt9WqscAQeA1YzlqgAAAAAAwJJDLx/S0QeOKjOdkWu5iiViisajkimFfijf9eVZ3tIMjjAM9dipx9pWf63B2PJB0eXX1/PX8a2Wa1Qr9SzfnHqlsrX+Ir/W+9UChEY0285a5VbSrc+tWr3V3q/2PWtEp/q01mfeaJ820y+tfoatfL8BrE3M5AAAAAAAAEtSe1J69DuPKnspq7l35paWrMrOZJdmc+Qu5pSZycjNuvr8i59Xcqrze3F0axmhflNtMLubddWqr9l2tlJnv6vW/l71KQCsRczkAAAAAAAA15i4b0JPFJ7Qic+d0JvPv6noYFRhEEqSDNOQZ3u68+E7616iqp7B1WYHYLtdrhf1NHqPVursRdlG99XohGaDg1bv26ny3erTZsr2qk8ArF2EHAAAAAAAYEUHnjugA88d0Ozrs0q/lpZUnOmRvLvzMzcAAADqQcgBAAAAAACqSt6TVPIegg0AANB/CDkAAAAAAKuSYRhLr8MwbPi6WuWrna+3bqw9jWwkzbI6AAB0HhuPAwAAAABWnVLIUAoYlocO9V4XhmHFgKJaOcMwlsqGYVixbgAAAHQeMzkAAAAAAKtSKYCoFTTUe1295ZqZuTFzekbp04t7WuxOaXxqvOF7oD8wOwMAgP5CyAEAAAAAQIPqDT2OHzyuMyfOKDYUkwxJi5e6lqtdD+3SwRcOdrilAAAAaxshBwAAAAAADSrf26M86Jg+Na0j+44osTmhbXu3KT4cl2EaCtxATtaRPWfr7Itn9dTAUzr08iGl9qS6/QgAAABrAiEHAAAAAABtNH1qWsc+c0zjU+Ma2zmmxNaEYkMxhUEoN+sqP5eXdclSfDiuzExGRx84qke/82ivmw0AALAqEXIAAAAAAFal0gyKWvts1HtdveVWmrmx3JF9RzQ+Na7xqXFtvn2zRiZGZMZMuTlX+St5DVwcUHQgKjNiSpIy0xk9u/dZ6bcbah4AAABEyAEAAAAAWIVK4UMpgKi0fFSt68pf11Ou2kbkxw8eV2JLQmM7x7T59s1KTiU1Ojkq3/GVu5BTdDAqwzAUeIF8x5dne3ItV/aCLee4I7FFBwAAQEMIOQAAAAAAq1Kl2RTl79d7XSPnK507c+KMtu3dpsTWhEYmRjQ6OaoN2zbInrPlO75cy5WTcRQfjhePRFyxREyDGwflnHGqtgcAAAAfZPa6AQAAAAAArAUzp2cUG4opPhxXbCgmM2bKd3zZc7Y821MYLM4OiRgyo6bMmCkzbioajyoaj0pRyZhtbEktAACA6x0hBwAAAABgVZmZmbnmkKSTJ08une/V65eee0kyJMM0ipuM51zlLuSUSWdkXbLkZB15tqfACxQGoRQuLpNlqhh8GKaUbrg7AAAArmssVwUAAAAAWDUOHz6sW2+9Vfv377/m/fI9M3rxuviGFLiB3Gxxk/HoYFSu5cowDHm2p8JCQW7OlWd78l2/GHj4YTH0AAAAQMOYyQEAAAAAWFXeeustpVKppUPSNaFHz14/UnztZB3l5/KyLlrKzmSVeS+jzExGuYs55a/kVVgoyMk5cq2ysCMMFaYIOwAAABrBTA4AAAAAANpgfGpcruXKnrNlXbIUHYgq8ALFh+Myo6bCIJRne3JyjgrzBTnZxaDD8op7dnihlOz1UwAAAKwuhBwAAAAAALTJrod26eyLZ4vBRqS48Xh8OC4zZkqh5Lu+XMuVky0GHaVZHfa8Le3qdesBAABWH0IOAAAAAADa5OALB/XUwFPKzGQkSZ7tKZ6Iy4ybMgxDgRfIsz25lqvCQkGF+UIx4AgkHZTEalUAAAANYU8OAAAAAADa6NDLh+RmXWWmM8qkM1pILyibziozk1F2NqvcxZysi5byV/KyrlgKw1CPnXqs180GAABYlQg5AAAAAABoo9SelB79zqPKXspq7p05ZdLFsCM7ky2GHOdzyl3MKTOTkZt19fkXP6/kFJtxAAAANIPlqgAAAAAAaLOJ+yb0ROEJnfjcCb35/JuKDkYVBsW1qAzTkGd7uvPhO/XgsQd73FIAAIDVjZADAAAAAIAOOfDcAR147oBmX59V+rW0pOJMj+TdzNwAAABoB0IOAAAAAAA6LHlPUsl7CDYAAADajZADAAAAAIA+YxjG0uswDBu+rlb5esrVqhsAAKAfsPE4AAAAAAB9pBQ0lAKG8uChnuvCMKwYUBiGsXQ+DMMVg41q5QFjA65HAAAgAElEQVQAAPoJMzkAAAAAAOgzpYBhpRCimes6aeb0jNKnF/cb2Z3S+NT4itdNPt3c/c89Xrvsucdr32ele1QrV6nOeupqRKv1NPpclcp1u75O1dnuz235/Wrdo9E+bfV73a3Pot3f0U6XA3D9IeQAAAAAAABL6l0q6/jB4zpz4oxiQzHJkLR4qWu52vXQLh184WCHW/q+yaebG6StVK7a4HPpXDsGXFutp9HnqlVnNbXa2u52NlNnuz+3evuq2T5tRbv7tBO/C534rgHASgg5AAAAAAC4jlSb9VEeapSWtlpu+tS0juw7osTmhLbt3ab4cFyGaShwAzlZR/acrbMvntVTA0/p0MuHlNqTklR7ILSewedqg7D1DNIuP1+rXK321hpMrlez9TT7XOVlGh1QrhQsdKqdK9VZft9a1zZaX7Pa1ae1NNunvfpdaLVfCD0A1MKeHAAAAAAAXGea3Xdj+tS0jn3mmManxrX9k9s1cd+EUrtTSt6d1JY7tmjTrZs0un1UYzvHlNia0NEHjuq9H77XoacoqneAuPy6egZlW6mvHu2op5HnKp1vdlC9kQHwWtfVuxRTpesabUs7n3mle/diKaVG+7SZcq30abP90qv+BLB6EXIAAAAAANBnSjMtau2zUe911co3EnQc2XdEm3Zs0vjUuFK7U7rxozdq/N5xbfnwFm3csVGjk6NaP7Few8lhjYyPKLYupmf3PttU29qh3oHy1faX4v36XOX19Ws769WPA+3N9ulq/ywAoBqWqwIAAAAAoI+UlpMqBRfLQ4jloUSt68pfl85X23OjPCxZfv74weNKbElobOeYNt++WcmppEYnR+U7vnIXcooORmUYhgIvkO/48mxPruXKXrD1zYe/qQe/8WDznYLrSjv3PWml/l7V14/hCgD0M0IOAAAAAAD6TKXZFeXv13tdO86dOXFG2/ZuU2JrQiMTIxqdHNWGbRtkz9nyHV+u5crJOIoPx4tHIq5YIqbBjYP60Qs/6ljI0esB6dVez2rQjhCg0U3Eexk09EMb6sF3FEC/IOQAAAAAAABVzZyeUWwopvhwXLGhmMyYKd/xZc/Z8mxPYbA4qyRiyIyaMmOmzLipaDxaPAajmn19Vsl7ki21o9qgarcHhLtVXz8MdJ97vNj3K20w3cmB7kr3biUEaHaD+07pZn92Sj98RwFc39iTAwAAAADWsNJyRvXs7bDSdbXKN1sOK7vttts0MzOzdJSUfj5//rxOnjy59H63XqdPpyVDMkxDYRDKzbnKXcgpk87IumTJyTrybE+BFygMQilcXPrKlIxIsUz6tXTT/dIvuvUX9v38l/ylsKN0dENpI+pmN6Sutz+7HTDU2jy9nwOPfv6OArj+MJMDAAAAANao5XsxlAKHlZYjqnbdSvs4LC9Xbb+ISuWwsieffFLpdHrFz2j5e716rVAK3EBu1lX+Sl7Rwahcy5VhGPJsT4WFgtycK8/25Lt+MfDww2Lo0SaVBlQrzTJop+s94Ki0MXWnN6yuFATU+5k3GnD0Q7+Xnq9f9VNfAYBEyAEAAAAAa1r5JtWtXrdSGbRPKpWq+f7WrVuXXu/fv78rr1O7i/U7WUf5ubwGLg7IMAw5GUdGpLjZuJtzVVgoyMk5cq1rww7DNJTas/KztUMjg97NuN4DjuX6uW3lmunPauHCavh8Oo0+ANCPCDkAAAAAAD119exVvfrlV/X2997WlbNXJEmbdmzS9p/Zrvv+xX0avWm0xy3E+NS4XMuVPWfLumQpOhBV4AWKD8dlRk2FQSjP9uTkHBXmC3Kyi0GH5cmzi0fy7tb24+gVAo7a+rHt/dim1Y4+BdCv2JMDAAAAANCySkth1fLib72or+75ql4/8rrsOVsbb9qoDR/aIOu8pdNfPa1n7nlG3/3t73agxWjUrod2FffguGgpO5tVJp0pHjMZZWezyl3MybpkKX81r8J8YWlWhz1v686H7+xJm2stpVRr0LZfA45Wn6tbmm1nq+1u5vnL9/1YaQ+QZvcEaVS12SSt9mm//y4AQDMIOQAAAAAALWk24Pjjn/1jvf6Hryt5d1I7/qsduvkTN2vivgkl70kqeW9SybuSGt46rB/8ux/o2M8d60DL0YiDLxyUa7lLoUYmnVHmvWLQkZ3NKnc+J+uipfzlvPJXikGHPW9LgfTgsQc72rZ69i8ov6ZWmX4NOCqVr/Rzu7QaqDTbzpWuq1bnahmMr9WfUvf6tF9+FwCgWSxXBQAAAABrWCmAqLXPRr3XVSrXqKMPHNXsG7Oa3D+psVvHNDw+rOhgVL7jqzBfkHXZkjViKTIYkRE1dO7lczr26WPSnoarQhsdevmQjj5wVJnpjFzLVSwRUzQelUwp9EP5ri/P8pZmcIRhqMdOPda2+msNxlbbpLpS+WoD5bXqbNeMg2bqaea5qtVTaUPxespW02w7a5VbSbc+t2r1Vnu/vN5mA6lO9Wm7fxfa3S/1fEcBXJ+YyQEAAAAAa1QpfCgFF8vDiOVhRq3rSu8vf738+tL75ecqlfu7E3+n6VemNXHfhG74yA1K7U7pxvtvVPLupMZ2jmnD5Aatn1ivxNaEElsSGhob0vD4sN556R3p71vvFzQvtSelR7/zqLKXspp7Z25pyarsTHZpNkfuYk6ZmYzcrKvPv/h5Jac6vxdHrWWEqg36rmbdfK5qddWqr9l2tlJnv6v1fe1FnwLAasVMDgAAAABYwyrNsih/v97r2nHue//qe8W9N7Zt0KZbNmnLR7ZobOeY8pfzyqQzCoNQvuPLzbtyLVdOzpGTdVRIFOR+15V2Vrw1umDivgk9UXhCJz53Qm8+/6aig1GFwWJQZhrybE93Pnxn3UtU1TO42o4B2Eb3Z+iGbj9Xq3X2omw/fG7NBget3rdT5bvVp73qFwDXH0IOAAAAAEDXZGeyunL2iib3TmrdxnUaGB1QPBGXJJlRU5GBiKKD0Q8e66KKJWLSu5KRa2xJLXTGgecO6MBzBzT7+qzSr6UlFWd6JO/u/MwNAACAEkIOAAAAAEDXpE+nl8IMI2Io8ALZc7ZkSIEXyLM9BV4ghYvLXJmGzIgpM1o8jIihMN34HiDonOQ9SSXvIdgAAAC9wZ4cqGml9XUbua5a+eXnap0HAAAAUJ+TJ0/q9OnT1xwlhw8f7unruX+Ykxkxi0tSFYqbjGdns8qkM7IuWCrMFeTmXHm2J9/1FfphcSmkxdAjYkSkK433CdANk0/XfwAAgPZgJgeqWr7xYClsWGlt3WrXlW9iWK7SWr3ldVWqGwAAAMC1XnrpJYVhqC9+8YsfOPfkk0/29PXozaPFGRt5T4WFgqxLlsyIKSfryIyY8h1fTra4B4drvR92BF6gwAvkB760qe6uAAAAwBpHyIGalocV1WZU1Htdp8389YwkaXxqvGdtAAAAALCy1L0pBX6gwkJB+St5xYZiCoNQ8UxcZtRU6Idy866cjFM8co48yyuGHY6vMAgVpvjjJ/QnNkwGAKD7CDnQc8sDkWZnavzgd36gV37vFc2dm9PAyIAkqZApaHRyVPf/xv366OMfbUtbAQAAALRmeHxYYzvGlL+UV25DTmbUlO/6ig/HFYlFistYOb6cnCNnYfFYnNXh5l1pTFKi108BAACAfkHIgZ4qDzXKl7mqNSPEyTp6ZuoZ2ZdtrduyTpN7J2VEDQVuICfnKHchp5eefEmn/+C0vnD6C4oNxTr2LAAAAADq8/EvfVwnHjmh2EhMhmHIK3hLIYckBW4g13blZB0VFgqyF2w5WUde3pN+sceNBwAAQF8h5EBfK9+TYzn7qq0v3/FlxRNxbfvpbRpODhenuoehXMtVYa6goc1Dsi5amn93Xr93y+/p1/6/X+v2IwAAAAAoc9vP36ab9t2kc395TmFQXJ4qnogrEo9IhhT6xdkcrrUYdMwXZF22dMvP3qK/v/XvJVarAgAAwCJCDtRUml1Ra1ZFvde10oblvrLrKxoYHdCN99+osdvGNJIaUSQekWd7sq/asi5Zig5FFYkW/6E0/+68fv8jvy/9ctubBwAAAKBBj/zHR/Tvf+7f69x/OifP8hRLxBSJR2SYxf/vHzjF2RxuzpU9b+uWT9yih//Dwzp8+HCvmw4AAIA+QsiBqkqhRSm4KJ9ZUb60VKXryl+XzpcHIpXKlQccL/7mi/JsTx/6xx/Sll1btPWurdqwbYNCL5R1xVJutri2bxiGCrxAnuPJt33NnZuTvivp4013CQAAAIA2efTbj+q7//N39dpXXlP2fFaRgYiiA8V/pnq2J7/gK7Iuop/6zZ/S/sP7e9tYAAAA9CVCDtRUaTPw8vfrva7Vc6Ef6vTXTmvzbZu1PrVeG7Zt0NitY9p480blr+QlQ/IL709td0YcOcPFY2B0QO6rroyfaf9sEwAAAACN+8T/+gnt/sJu/fArP9Q7331Hl398WZK0aecm3fyJm3Xfr9+n9Teu73ErAQAA0K8IObDqXP7xZQVOoMGNg4qPxBVbV9ys0M25Cv1QhmnIjJoyY6Yi8UjxGCgescGYDN9QcCXo9WMAAAAAWLRhcoN+9n/72V43AwAAAKuQ2esGoH/Nz89fcyx//yc/+cnSzz/+8Y+7+nrm9IxkSJFYRFJxGrt1yVImnZF1xZKTc+Q7vsIgLM4GMSTDNGREiuGHYRhSuvH+AAAAAAAAAAD0F0IOrOhrX/uaTp8+rYsXLy4dJRcvXtQ//MM/LP386quvdvW1fdWWYRry3eKSVPmreWVns8qkM8qdz8m+YsvJOHItV37BV+AGCvxACopLYJkyZdgsVwUAAAAAAAAAqx3LVWFF6XRa7733nr74xS9+4NyOHTu0Y8eOpZ8feeSRrr5+9z+9qzAI5Vqu7Hlb1kVLhmnIyToyIoYCN5CTc1RYKMjJOfLyxQ0LfddX4AXyQ19KNtIbAAAAAAAAAIB+RMiBVSe1OyWv4KkwX1D+cl7RwaiCIJCTcWRGTQV+IM/25GTfDzpcy5VnF8OO0AsVpipveA4AAAAAAAAAWB0IObDqRNdFNfnTk7rw/15QfCQuM2IqcAMVEgVFYhGFYajACeTmXRUWCrLnbTlZpzi7I1OQtkuK9PopAAAAAAAAAACtIuTAqnTw+YP63YnfVe58TpLkFTzFEjFF4sX0IvCKsznc3LKgY8GRZ3vSQ71sOQAAAAAAAACgXdh4HKtSYktCD/zuA8qezyo7U9x0vHRkZ7LKnc/JumDJumQpfyUv+6otJ+foU//np6R1vW49AAAAAAAAAKAdmMmBVWvPr+1RJBbRtx//tgoLBQ1sGFAkHpFhGlIg+Z5f3JsjU5zB8akvf0r3HLpHf374z3vddAAAAAAAAABAGxByYFWb+sKUPrT3Q3r+F55XJp2R7/iKxCJSWAw5IvGI1qfW63N/9jlt2rmp180FAAAAAAAAALQRIQdWvS13bNGv//2vK/1aWj95+Sc69/I5SdLk3klt+8fbNH7veI9bCAAAAAAAAADoBEIOrBmp3Smldqd0/39/f6+bAgAAAAAAAADoAkIOAAAAAMCqZBjG0uswDBu+rlr5es/VqhsAAACdZfa6AQAAAAAANKoUNJQChvLgoZ7rwjCsGFBUO7f8PAEHAABAbzGTAwAAAACwKpUChjAMK4YcjVzXSTOnZ5Q+nZZUXGp3fGrlvQMnn27u/ucer1323OOVz1UqW61MrfK1yrZaZz168Vwrlet2fZ2ss52f2/J7Nfp9afb71Ur5TnwW7f6ONvv7U89n0WqfAkCnEHIAAAAAANCgepfKOn7wuM6cOKPYUEwyJC1e6lqudj20SwdfONjhlr5v8umVByGrDVyWzjUz6FmpvlbrrFerdVQq3+xzVdNsP1Yr22y5amXb+bnV21fN9mkr2t2n3f7da1Qv+hgA2omQAwAAAACABqy0R0f5e9OnpnVk3xElNie0be82xYfjMkxDgRvIyTqy52ydffGsnhp4SodePqTUnpSk2gOa9QwgVxtMrTTYWqveWoO75eeaLddI2Xo0+1yV/qq90edqdPC40X5stZ0r1Vl+33qub7TOZrSrT2tptk+bLdeu371uBhXM2ADQb9iTAwAAAACANpo+Na1jnzmm8alxbf/kdk3cN6HU7pSSdye15Y4t2nTrJo1uH9XYzjEltiZ09IGjeu+H73W0TY0GDfWcqxa81DOwXumadg2gNvtc1a5r5bkqaaUfK11X71JMjX4G7ejTeuovv28vBtUb7dNmyrXSn+3ql3bNnAKAXiLkAAAAAACsSqUlo2rts1Hvde1yZN8RbdqxSeNT40rtTunGj96o8XvHteXDW7Rxx0aNTo5q/cR6DSeHNTI+oti6mJ7d+2xX2tYLq20pnHoDgG4/V3l9/drOevXjoHqzfcpnAQC9xXJVAAAAAIBVp7SJeCm4WL5c1PLlo2pdV/66dL7ec+X3PH7wuBJbEhrbOabNt29Wciqp0clR+Y6v3IWcooNRGYahwAvkO74825NrubIXbH3z4W/qwW882GLPANX1+i/3ex0OMaD/vnbtIUOfAug1Qg4AAAAAwKpUacPv8vfrva4d586cOKNte7cpsTWhkYkRjU6OasO2DbLnbPmOL9dy5WQcxYfjxSMRVywR0+DGQf3ohR91LOToxIbYndKNQfh+/Yv6XmjXgHU9fdrrgKVf2lCPTn9H29kPq6VPAaxdhBwAAAAAALTBzOkZxYZiig/HFRuKyYyZ8h1f9pwtz/YUBouzSiKGzKgpM2bKjJuKxqPFYzCq2ddnlbwn2VI7mtk0upZK+0VMPr3yxsiNDNCudG23Bkv7YVC2Xf3YqEr3bnXAupmNzjulm/3ZKZ3sr2buvRb6FMDaw54cAAAAAIBV5bbbbtPMzMzSIUlvvPHG0vmTJ0/25PVLz70kGZJhGgqDUG7OVe5CTpl0RtYlS07WkWd7CrxAYRBK4eLSV6ZkRIpl0q+lG+6PTmlksLs0SF862lV3J/TzX523ux/rUdrAupWNrOvp024PhtfayL2fB+c7/R1t9tlXc58CWNuYyQEAAAAAWDWefPJJpdPpqktS9ep18Q0pcAO5WVf5K3lFB6NyLVeGYcizPRUWCnJzrjzbk+/6xcDDD4uhR5tUGhitNFug0rXV7lVeV6Ulj+oZ9Fypjkba2oh+DTja0Y+t1Fv+Xru/K/3U76Xn61fdCjjaef9+71MAax8hBwAAAABgVUmlUh947+677156vX///t68fmS/3v79t+VkHeXn8hq4OCDDMORkHBmR4mbjbs5VYaEgJ+fIta4NOwzTUGrPB5+tXeodvG5mELTdA7KNDrTXo58G2ivp57atpNE+rTYQvho+n07rZh/wWQBYSwg5AAAAAABog/GpcbmWK3vOlnXJUnQgqsALFB+Oy4yaCoNQnu3JyTkqzBfkZBeDDsuTZxeP5N2t7cfRqrW6GXE/taVR/dr2fm3XakV/AkDzCDkAAAAAAGiTXQ/t0tkXzxaDjUhx4/H4cFxmzJRCyXd9uZYrJ1sMOkqzOux5W3c+fGdP275WB1kbfa5as0j6pZ+abWc7lhZqpk/bda9WVXv2Vvu02e9MN/ugE58FS1UB6DU2HgcAAAAAoE0OvnBQruUqM5NRdjarTDqjzHsZZdLFn3Pnc7IuWspfzit/Ja/CfEH2vC0F0oPHHuxo2zqxPE2le9Y7qNvofRvV6uBxeTs6NZjbbD9WKl9vO1e6rp8G5JtVqz+l7vVpvd/1fu5PqT19CgCdwkwOAAAAANc9wzCWXn9gI+k6rqun/ErXLH+vnvqxOhx6+ZCOPnBUmemMXMtVLBFTNB6VTCn0Q/muL8/ylmZwhGGox0491rb6aw2qlg9ELr++1l+5N1NfJc1uSt7M/Rt9ruWzHVYq22hfVNpQvJ6y1TTbzlrlKmn1u9KMZvu02e9lp/q01mfeaH+28l1rFjM2APQrZnIAAAAAuK6VgoZqwUOt68IwrBmOlK5Zft3y9wg31o7UnpQe/c6jyl7Kau6dueJsjnRG2Zns0myO3MWcMjMZuVlXn3/x80pOdX4vjnOPd2aD8GbqqtWOTrS1UdWerZt11dNXjbzfjjr7Xa3vXi/6dLVrtU8BoJOYyQEAAADgulcKGMIwrBhyNHLdcqWAo+XrQumdk+/o7e++relXpiVJN37sRm3/me26ad9NUn3NQZdM3DehJwpP6MTnTujN599UdDCqMFgMyExDnu3pzofvrHuJqnoGEJsdZGx1cLJX9Xbj/o3eo5U6e1G2m8/X7D3X8ve6V9+XZu9JkAGgXxFyAAAAAECH1bscViU/+auf6E8+9yey52wZpqH4UFxhEGr6lWm98juvaHDToD77wmeVuj/VzmajDQ48d0AHnjug2ddnlX4tLak40yN5d+dnbgAAAFwPCDkAAAAAoMPK9+8oDzqqzeL4q//9r/SX//ovNXrzqLbeuVXx4bgMw5BX8ORkHOXn8sqdz+nIx4/ok1/6pO5//P6OPguak7wnqeQ9BBsAAADtRsgBAAAAAH3q5L8+qVd+5xXd+LEbtWnHJg0nhxVbF1PgBSpkCspfyWvg0oBi62KKrovqe//L9+Rabq+bDXREI5ses6wOAADXD0IOAAAAANe90kyKWvts1HtdO8y9PaeX/83L+tDHPqSt/2irtnx4i9ZPrJcRMeRkHFmXLcUTcUViEUlSGIQKvEAvHX5J+jVJIx1vIgAAANBzhBwAAAAArmul0KIUXFRaWqrWdeWvK21SXu9SVX/6T/9UGz60QaPbRzW2c0zJu5LaePNGOVlH2fNZGVFDoR/Kd3x5ticv78nNuXLmHFnftKTPt9QtQN9hdgYAAFiJ2esGAAAAAECvhWG4dJS/X+915Uc95VaqQ5KcrKPpV6Y1cuOIhjYPaWjLkBI3JDS4cVDxkbhiiZjiQ3HFhmLvH+tiig5FFVsfk96VDK/zs00AAACAXmMmBwAAAAD0mZnTM4oMRIrBxUBUhgwVsgXlzufkO74CNyiGI4ZkmIaMiCEzZioSiygSi8iIGAreC3r9GAAAAEDHMZMDAAAAwHXp3Llz+tu//Vu99dZbS0fJW2+9pfPnz+uP/uiPlt7r5utvfe1bMk1TMiTf9VXIFAOOTDqj3KWcCvMFuZZbDDz8QGEQXhN6mKYp4zwzOQAAALD2MZMDAAAAwHXp61//ujZt2qRf+qVf+sC58fFxxWIx7du3b+m9br7+yH0f0ff/9PvybE9OxlH+cl6ReERuzpVhGvIKxfedrCMv770/u8MLFfqLS2MNfXAZLAAAAGCtIeQAAAAAcN26fPmyRkZGPvB+6b2bbrpp6b1uvr7r5+7Sqf/pVDHguJJXfDiuMAzljDjFpai8QF7eU2GhoEKmIDfnFjcfL3jyXV9BEMiYYCYHAAAA1j5CDgAAAADoM6PbRxUfjsu+Ysu6ZCkSjyjwAhUyBUWiEYVBKK/gyc25S0GHk3Pk5l25eVcakDTa66cAAAAAOo+QAwAAAAD6jBk1tedX9+j7v/N9xYZjMkxDvuMrlogpEo9IYXGvDi/vyck6KswXVFgoyMk6crOu9F9IocFyVQAAAFj7CDkAAAAAoA/tP7xfb/xfb2hhekEKJc/2lkIOw1hcsqrgybVcOZli0JG/ktdIakRXfvqKRMYBAACA64DZ6wYAAAAAAFb2q2/+qiLxiObemVMmnXn/mMkoez6r3IWcrIuW8pfzyl7IamD9gP7Z3/6zXjcbAAAA6BpmcgAAAABAn4on4vqNd39DX7v/a7ry4ysyTKM4myMaURiGCpxAjuVIoXTDrhv0y6/8ssR+4wAAALiOEHIAAACg6wzj/VHYMKy8pk6l62qVr3a+3rqBfmFEDH3htS/oja+/oVP/9pQuv3VZZqw4KT9wA43tHNPHfvNjuuuf3tXjlgIAAADdR8gBAACAriqFDGEYyjAMGYZRNahY6brSf5cHFsvLlQciy6+vdA7od3f90l2665eKQcaVH1+RJG26dVMvmwQAAAD0HCEHAAAAum55WLFSUNHodSuVaZf85bzSp9OSpNTulNZtWtfW+wPNINwAAAAAigg5AAAAcN0oD0uqBSLf/R+/q9e+9pqcnKPYYEyS5OU9xYZj2v3f7tYn/s0nOt5eAAAAAEB1hBwAAABYs2otT7XSclUL7y7omd3PSIE0um1U68bWyYgYCtxATs5R/lJer37lVf3N1/9GX3jtCxqZGOna8wAAAAAArkXIAQAAgDWpmf025t6Z0x/e/4dKbEloy64tGk4OKzYUUxiEci1X9pyt/Ka8Bi8OauG9BT0z9Yx+5dVf6dATAAAAAABqIeQAAABA15UCiFr7bNR7XaVyjfqDu/9Ao9tGldqT0ubbN2t4fFiReERe3lP+al7WRUuxwZjMmCkZ0sJPFvTMPc9I/13DVQEAAAAA2oCQAwAAAF1VCi1KwUWl5aNqXVf+unS+9PNKe29U25PjW7/8LUWiEW3ZtUVbPrxFybuT2rBtg3zPV/5SXrHZmAzTUBiE8l1fnu3Jsz3NvTMn/bmk/7r1vgEAAAAANIaQAwAAAF1XaZZF+fv1XlfvuWrn/+brf6OJ+yY0PD6s9Teu18ZbNmr0plHZV+3iclV5V07OkZN1FB+JKz4cVzwR17rRdXL/2iXkAPpMpUCzmetW2s+nWpl66wYAAEDrzF43AAAAAOi1C29eUDQe1eCGQcWH44oORhUGoZyMI9/1ZciQGTFlRk1FYhFF4hFFBhaPwYiMiCFd7PVTACipNMOrmetWei8Mw5qBSOlodLk9AAAANIaZHAAAAOiay5cvX/Pz2NiYJOmNN97QXXfd1bPXM6dnJFPFsCKU3Lwr66KlwA0UBIGcrCPP8RT64TWDoYZZDD8Mw5CRZiAT6CeVlqlr5Lpm9wVq1MzpGaVPpyVJqd0pjU+Nr3jd5NPN3f/c47XLnnu88rlKZauVqece9ZRvtlw9Wn2udj1Tt+vrZJ3t+K6sdK9a5Rvt0wj++X8AACAASURBVFZ+HyqV78Rn0e7vaKOfQyPtbednDwCNYiYHAAAAuuLpp5/WD37wA2UymaWj5O233+7pa8dyZMhQ4AVyco7yV/PKzma1kF5Q7nxO+at5OQuOXMuVX/CL4YcfKAwW/1JbhkKHJWmAtaR8iap6Ld9PqNY9jh88rsPGYR356SP6i//hL/QXv/EXOrL3iA4bh/Un/82ftNL8hlUaoKw2GDz5dO3B4mrXNHvvZsOedtRdq3yr9220XCfa2Wyd7Xz2eq9ttk9b0a3PopX+bLVfGv38O9kWAKgHMzkAAADQFXNzc/rhD3+oT33qUx849wu/8As9fT1+97jCMJSbc2XP2bIuWDIMQ/FMXGbUlO/6cnOuCgsFOVlHbt6Vb/sKnECBF8gPfRkpZnIAa0WzAcdKZVe61/SpaR3Zd0SJzQlt27tN8eG4DNNQ4BZnjtlzts6+eFZPDTylQy8fUmpPSlLtv6Cu5y+mV7qmVH7y6ZXP16q3Url6622kXCN1NnrveuuoNMOg3raVzjU68NtoX7TazpXqLL9vPdc3Wmcz2tWntTTbp82Wa7U/m+2XavU3+vl3+rMHAImZHAAAAIBSu1Ny7WLAkb+cV/Z8Vpl0ZunInc/JumQpfzVfDDoyjhzLkWsXww65UphiJgewliyfjVH6uR2mT03r2GeOaXxqXNs/uV0T900otTul5N1Jbbljizbdukmj20c1tnNMia0JHX3gqN774XttqbuSegZJGz0n1Q5fmglt2jVI2spzVbqunsCg2UH1Vvqi0XY2+7m1o0/rqb/8vr0YOG+0T5sp10p/ttIvzXz+1eoj2ADQDYQcAAAAuO6ZMVM7/ssdys5klbuQU242p2y6GHRkZ7LKzhbfz1/OXxN0lGZ3aKckJnIAfaXecGKl65ZvHL58z452OLLviDbt2KTxqXGldqd040dv1Pi949ry4S3auGOjRidHtX5ivYaTwxoZH1FsXUzP7n22LXWvNb1YAqfeAeBeL6HUr+2sVz8OjDfbp6v9swCA1YDlqgAAAABJn/3Tz+pLiS8pdyEnSfJsT7HhmCKxiGRIgRfIs71isJEpqDBfKIYdliM93OPGA7jG8n0xSj+XLF8+qtp11SwPRMrLlm9Uvvyexw8eV2JLQmM7x7T59s1KTiU1Ojkq3/GVu5BTdDAqwyjuD+Q7fvF/cyxX9oKtbz78TT34jQeb6Y6eaOSv8dE/ev259Tocut6/r73+/AGgWYQcAAAAgKToYFQPfeMhfetXvqXQD+XlF0OOeKQ4KOqH8hxPXt6Tk3VUyBTk2Z4++83P6vk3npdYrQroK5UCi/L36wk2Gi1T6fyZE2e0be82JbYmNDIxotHJUW3YtkH2nC3f8eVarpyMo/hwvHgk4oolYhrcOKgfvfCjjoUczQ4st7KRdD8PovIX9e9r1+dWT5/2wwB7P7ShHt36jrbj818tfQpgdSPkAAAAABbd8dAdMiKGjj98XIWFggY2DCgyEJFhGlIg+V7xL6sLCwW5eVefPfFZ3fbzt0lv9LrlAPrdzOkZxYZiig/HFRuKyYyZ8h1f9pwtz/YUBouzSiKGzKgpM2bKjJuKxqPFYzCq2ddnlbwn2VI7mtk0upZq5SrVV2uvicmnV96ouJsBRD8MyvaqL5r53OrRzEbnndLL71a7dKq/Wvn8VyrbD79LANY2Qg4AAABgmdsfvF3/8vy/1LFPHdPs38wuLT0TKpQCyTANJaeS+iff/icaWD/Q6+YCKDMzM3PNz+Pj402//+677+r++++XJJ08eVL79u1r+vXw3w9LRvF/Q8IglJtzlbuQk+/4CoNQTtaRZ3sKvEBhEErh4lJYpmREimXSr6VbDjnapdHB7mYHlHu1fFE/Dsr2YhC+HUFAswPjnVQtXCu1px+/A1J3v6PtCoL6uT8BrA2EHAAAAECZdRvX6bHvP6aF6QW994P3dO4vz0mSJvdNauK+Ca2/cX2PWwhgJYcPH9att96q/fv3f+BcvctXVXp/+c9Nvw6lwA3kZl3lr+QVHYzKtVwZhvH+LLGcK8/25Lt+MfDww2Lo0SaVBhorzRaodG21e9VTZ7UZCsvLVFoupxOD4v0acPSiL5bfv/y9dn9X+qnflwcd/aiXAUfpvVqf/0rvN/KdAYBmEHIAAAAAFay/cb3W37hedxy4o9dNAVCnt956S4888sgH3k+lUiteX+395eeWByfNvJ5ZX5wx4mQd5efyGrg4IMMw5GQcGZHiZuNuzlVhoSAn58i1rg07DNNQas/KbW2Hegevuz0g3a16+mmgvZJ+bttKGu3TauHCavh8Om0190Gj4RgANIqQAwAAAACADhufGpdrubLnbFmXLEUHogq8QPHhuMyoqTAI5dmenJyjwnxBTnYx6LA8eXbxSN7d26Wq+mWQtd3t6Jfnaka/tr1f27Va0Z8AUJ3Z6wYAAAAAAHA92PXQLmXSGVkXLWVns8qkM8VjJqPsbFa5izlZlyzlr+ZVmC8szeqw523d+fCdPW17O5ao6kfNPlenNuZul2bb2Y52N9OnlY7yazqt2mySVvu02e9Mr5eoAoDVgJADAAAAAIAuOPjCQbmWuxRqZNIZZd4rBh3Z2axy53OyLlrKX84rf6UYdNjzthRIDx57sKNt6+RSQSvdu96B3Xa3pZ33Km9jp/ZyaLUvmm1nK59bPw+W1+pPqXt9WqtcL/uzkc+/3ucAgE5huSoAAAAAALrk0MuHdPSBo8pMZ+RarmKJmKLxqGRKoR/Kd315lrc0gyMMQz126rG21V9rsLHa4GU9f+Ve/l6pTDODnJ0cGO3kczUb3FQr30xfNNvOZj+3Vvq0Wc32abPfrU71aa3PvNH+bOW71o7Pv5J+Dr8ArG7M5AAAAAAAoEtSe1J69DuPKnspq7l35paWrMrOZJdmc+Qu5pSZycjNuvr8i59Xcqrze3F0ajmgaoO+1eprtly3VGtfN+uqVV+z7ez3/m9Fre9dL/q03zT6+dfz7P34nADWDmZyAAAAAADQRRP3TeiJwhM68bkTevP5NxUdjCoMQkmSYRrybE93Pnxn3UtU1TN42OwAYzsGJntZdyfv3eg9WqmzF2W7+XzN3rNX361u1Nur70uz9yDEANBLhBwAAAAAAPTAgecO6MBzBzT7+qzSr6UlFWd6JO/u/MwNAACAtYKQAwAAAACAHkrek1TyHoINAACAZhByAAAAAMAaZhjG0uswDJu6rtY96jnf6D2BRjSyQTLL6gAAsLaw8TgAAAAArFGlIKEUIiwPFuq9rtY9SgFG6ah072plKrULAAAAqIWZHAAAAACwhpWCh1phQrXrKp1baYbGcqXzle7XiJnTM0qfXty3YndK41PjDd8DaxezMwAAuH4RcgAAAAAAmlZp2al6ApCVypU7fvC4zpw4o9hQTDIkLV7qWq52PbRLB1842HzjAQAAsOoRcgAAAAAAmrZSsFEr4KhUbrnpU9M6su+IEpsT2rZ3m+LDcRmmocAN5GQd2XO2zr54Vk8NPKVDLx9Sak+qvQ8GAACAVYGQAwAAAADQdpX27qjH9KlpHfvMMY1PjWts55gSWxOKDcUUBqHcrKv8XF7WJUvx4bgyMxkdfeCoHv3Oo514DAAAAPQ5Qg4AAAAAWMMq7YvRyHX13qOkPMwoDzhqBR5H9h3R+NS4xqfGtfn2zRqZGJEZM+XmXOWv5DVwcUDRgajMiClJykxn9OzeZ6Xfrqt5AAAAWEMIOQAAAABgjSoFE6VwolLQUO26es4t/7mRdq1U7vjB40psSWhs55g2375ZyamkRidH5Tu+chdyig5GZRiGAi+Q7/jybE+u5cpesOUcdyS26AAAALiuEHIAAAAAwBpWKXgof79aQNHsuWrXVCp35sQZbdu7TYmtCY1MjGh0clQbtm2QPWfLd3y5lisn4yg+HC8eibhiiZgGNw7KOePUbAsAAADWFrPXDQAAAAAAQJJmTs8oNhRTfDiu2FBMZsyU7/iy52x5tqcwWJxVEjFkRk2ZMVNm3FQ0HlU0HpWikjFb35JaAAAAWBuuCTlKU5DrXWcVAAAAANCfbrvtNs3MzGh2dlYnT55cer+fX7/03EuSIRmmUdxkPOcqdyGnTDoj65IlJ+vIsz0FXqAwCKVwcYNzU8XgwzCldI2OAQAAwJqytFzV8vVVS0FHveupAgAAAAD6x5NPPql0Or3077vyfTT69XXxDSlwA7nZ4ibj0cGoXMuVYRjybE+FhYLcnCvP9uS7fjHw8MNi6AEAAIDrzjV7cpRvOgcAAAAAWJ1SqdTS62QyufR6//79/fv6kf16+/fflpN1lJ/La+DigAzDkJNxZESKm427OVeFhYKcnCPXKgs7wlBhirADAADgetLUxuNXz17Vq19+VW9/721dOXtFkrRpxyZt/5ntuu9f3KfRm0bb2kgAAAAAwNo3PjUu13Jlz9myLlmKDkQVeIHiw3GZUVNhEMqzPTk5R4X5gpzsYtBhecU9O7xQStauBwAAAGtHwyHHi7/1ov76D/9aCqXB0UFtvGmjAj+Qdd7S6a+e1uvPvq7dv7pbn/jSJzrRXgAAAADAGrbroV06++LZYrARKW48Hh+Oy4yZUij5ri/XcuVki0FHaVaHPW9Lu3rdegAAAHRbQyHHH//sH2vm9IySdyc1vHVY8URckuTaxenC9pXiX9v84N/9QOdfP69Hvv1IRxoNAAAAAFibDr5wUE8NPKXMTEaS5Nme4om4zLgpwyguWeXZnlyr+O/QwnyhGHAEkg5KYrUqAACA68o1IUdpQ7qV9uM4+sBRzb4xq8n9kxq7dUzD48OKDkblO74K8wVZly1ZI5YigxEZUUPnXj6nY58+Ju3p2rMAAAAAANaAQy8f0tEHjioznZFruYolYorGo5IphX4o3/XlWd7SDI4wDPXYqcf0zJ890+umAwAAoMuWQo5SuFEKOEqbkEvS3534O02/Mq1te7fpho/coK13btX6G9dLhmTP2cpdyCk6GJVhGgqDUIEbyHd8vfPSO9J6STu7/VgAAAAAgNUqtSelR7/zqJ7d+6zsBVuDGwcVjUdlRBb/zbk4m6M0g+OxU48pOZWU/qzXLQcAAEC3XTOTY3mwsdz3/tX3tPGmjdqwbYM23bJJWz6yRWM7x5S/nFcmnVEYhPIdX27eLa6NmnOK66MmCnK/6xJyAAAAAGvA8hnflf7tUO26WuXrKVdP/VgbJu6b0BOFJ3Ticyf05vNvKjoYVRgUP3fDNOTZnu58+E49eOzBHrcUAAAAvVRzT47sTFZXzl7R5N5Jrdu4TgOjA0t7cZhRU5GBiKKD0Q8e66KKJWLSu5KR++A/SgAAAACsHstnfJdmgFcLKla6rvTflUKL8vutVK68DlwfDjx3QAeeO6DZ12eVfi0tqTjTI3l3ssctAwAAQD+oGXKkT6eXwgwjUtzkzZ6zJUNLU4QDL5DC4j82DNOQGTFlRouHETEUpvkrKwAAAGC1Wx46VAsa6r1upTK1VApXVuI7viQpEo/UdT36W/KepJL3EGwAAADgWlFJOnz4sJ588kmt9PrnNv6czIhZXJKqUNxkPDublZt3ZciQa7lyc64825Pv+gr9sDiFeDH0iBgReVe83j0hAAAAgOvG2//P2/r+v/2+0q+lZV2yJElDW4aUujelj/3Wx3TTvpt620B0TavLq1U7Vx7esXwaAABA70Ql6dOf/vTSG6WAo/T6rT9/qzhjI++psFCQdcmSGTHlZB2ZEVO+48vJFvfgcK33w47ACxR4gfzAlzZ1/8EAAAAArE7VlsKqNpj8jV/4ht59+V1F4hEN3TCkjbdsVOAGsq/amv7+tJ7/xed188dv1kN/8lAnm48+0I7l1aotoVYqAwAAgN6LStK9995b8YLUvSkFfqDCQkH5K3nFhmIKg1DxTFxm1FToh3LzrpyMUzxyjjzLK4Ydjq8wCBWm+D9/AAAAAGprZDmqkjAI9eUPf1n2VVsTuyeUSCaK+wiGkpt3i/+WuZxX7lJO//n//s/6yke+on/+o3/eoSdAv2h1ebV2hxgzp2eUPr24p8julManxle8bvLp5u5/7vHaZc89XvlcpbLVytQqX6tsq3XWoxfPtVK5btfXyTrb+bktv1ej35dmv1+tlO/EZ9Hu72izvz+NfBbtKAcA7VRzT47h8WGN7RhT/lJeuQ05mVFTvusrPhxXJBYpLmPl+HJyjpyFxWNxVoebd6UxSYkuPAkAAACAjioFELX22aj3ukrlGvXVe78qN+vqpp+5SWM7xzQyPqLIQER+wZc9b8u6ZCmWiCkyEJEZMTX3zpy+dt/XpE/XvDVQUb2ByPGDx3XmxBnFhmKSIWnxUtdyteuhXTr4wsEOt/R9k0+vPAhZbTC4dK7S4GWtss2Wq1ZnvVqto1L5Zp+rmmb7sVrZZstVK9vOz63evmq2T1vR7j7t9u9eozrxvQWAbqoZckjSx7/0cZ145IRiIzEZhiGv4C2FHJIUuIFc25WTdVRYKMhesOVkHXl5T/rFjrYfAAAAQBcsX86n9HPJ8nCi1nXlr5cvDVR+TbWlgkpe/fKruvr2VW3fv1037LpBW//RVq3/0HoplPJX88qdzykSjxTL+6F815dX8HTx7y5K45KmWuoWXCdqLVW10vdz+tS0juw7osTmhLbt3ab4cFyGaShwAzlZR/acrbMvntVTA0/p0MuHlNqTklR7QLOeAeRqg6mVBltr1VtrcLf8XD3lWq2zHs3WUemv0xvtj0YHgRvtx1bbuVKd5fet5/pG62xGu/q0lmb7tNly7frdI3AAcD0z67notp+/TTftu0nz5+aVmckok84o897if2cyyp7PyrpoKX85L/uqrcJcQdZlSzd/8mbp1k4/AgAAAIBuCMNw6Sh/v97ryo96zq1Uh1RcpurU/3FKm27ZpPUfWq/R7aPafMdmjU+Na8PkBiVuSGjd2DoNjg5qYP1A8RhZPIYHpJclQ43NNsH1p5kZRtOnpnXsM8c0PjWu7Z/cron7JpTanVLy7qS23LFFm27dpNHtoxrbOabE1oSOPnBU7/3wvQ49QVEjAUW956oFL80MrNdbtl7tqKP8unqeq9lB9Wb6sdJ19S7F1GgA1a7PrdHQrhfLIDXap82Ua6U/29Uvzc6cateMKwBoh7pCDkl65D8+om3/eJvm351XNp0tBhzpjLIzWWXPZ5U7n1PuYk7WJUu5Sznd8olb9PB/eLiTbQcAAABwHVv4yYKy57Ma2jykgQ3F8CI6EJVf8CVDMqOmIvFI8RiIKDoYXTpiQzEpI2mh10+BTllpdlCj1zW7hNqRfUe0accmjU+NK7U7pRs/eqPG7x3Xlg9v0cYdGzU6Oar1E+s1nBzWyPiIYutienbvsw3Xs1qstr8wrzcA6PZzldfXr+2sVz8Ojjfbp9frZ9GPnyGA61PdIYckPfrtR3X/4/fLnrd19R+u6urbV7UwvaDMexnN/2RememMXNvVT/3mT+nhPyPgAAAAANaCoaEhOY6zdJTMz8/39HX6tbSMiCEzZsowDfmOL+uKpUw6I/uqLS/vKXADhcHiILUhGaZRLBM1ZZqmwvfau7k0+kP5MmiVlk2r57rS8mvly60tP5aXO37wuBJbEhrbOabNt29WciqpifsmdMOuGzR60+j/z979xshxHnae/1VNd8+QPUMOyaHZ3aOQJ4SUZNN0yDEpeRfHFXfjjYzcOgjIEUJLd6cFBSNBgF3l7nA4YO8EQYDuxe3u3UGHjfNisyIWx6NsEtQCyWbPWC0CMxB4WZ9oJSuZRhTzbDlU94ik5Znpf9VV1V33otmjUav/VFdXdVfPfD9AQ82qeup56qkavXh+/dSjudyc0p9La+fCTu3Y25xpNLN3RolUQm+cfyPsrgA+Y9y/wB9HOLT5g0+wFgeArWKgkEOSfvV//lX9zl/8jh7/x49r3+F9ci1XruVq7yN79cTvPaHfffd3deblMxE0FQAAAMCo/eZv/qZ+9Vd/VX/1V3+18Wm5ePHiWL+vf7guc8qUV/fkVl1Zq5bKK2UV80WV75ZVXa021wq0mmFHw30QeDwYjzYNU8Y6r6vaqkb5erXNbl27pT2H9yh9IK25xTnNH5rX7oO7tXNh58ar01KzqU8+6ZSS6aRm9szo3SvvhtwLn5ikwcxR1Mkg7SfCCgH8lB13wNJqwyTc/6jbyGuqAGwlvhYeb7f70G79/f/l74fdFgAAAAAx8yu/8itd9/3e7/3eWL8vPLaghtuQU3VUW6+pcr8iY8qQXbJlTDVndthlW3bRll1uhh31Wl11p65GvaF6oy5vPzM5EJ7CzYKSO5NKzaaU3JmUmTRVt+uyVi25liuv4TVnf7RmEyVNmSlTiVSi+ZlJaOWdFWVOZIZqR5BFo/vptl5Ea8C424LZwxjFIGocBmqj7sduup172EHsYdZjCdso+zMqUfYXr6kCsFUECjkAAAAAYNxyJ3NquA3V1mqq/LyixI6EvIYnu2jLTJhq1Btyq67sUjPocMqOnKqzEXZ4dU/GIjM5tppHH31UhUJh49/ZbFaSPrWttf3HP/6xDh8+LEm6fv26nnzyyaG+52/mN16L5jU8OWVH5btl1e1689lszSzaNKvIMAzJlIypZpn82/mhQ46wDDLYHdbg8Sh+JR7nX6KPYxA+jCDAT5+O+tp6BXOt9sTxGZCif0YnaWYXAPhByAEAAABgIu3Yu0MHvnRApUJJ07umNZWYUsNpKDWbkpk05TU81e26nEpzpkdtvSa7ZMupOHLKjpSVvGlmcmwlL730kvL5fMfFwvttC+u7PKnhNOSUHFU/rioxk5BTcWQYhlzLVW29Jqf8IGxz6s3Ao+59snZMCLoNjHabLdDt2F7naq+rffAzyELL2zngCLMfg9Tbvi3sZyVO/b456IijUQUcvKYKwFZCyAEAAABgYv367/+6/vXf/ddK3U1JhuTWXCXTSU2lpiRJDffBbI6y3Qw61mqyi7bcmiv91pgbj0jkcjnf248cObLx/cyZM0N/z51s1mGXbFVXq5q+Ny3DMGQXm69Qa7gNOeVm6GaXm4Hb5rDDMA3lTnVufxj8Dl4HGcwcduBzOwccm8W5bZ0M2qe9woVJuD9RG2UfBL0X3EMAcUTIAQAAAGBiLT6+qC9+44v64bd/KK/RXIC8FXIYhtFce8Ouy600X1tlrVuyfmHpS//1l3Qze3NjEXIgDNmlrJyKI2vVUuV+RYnphBrug9lFiebsItd6ELqtfTKzyK24cq3mJ3N8vK+qCnOQ0u+5CDh6i2vb49quSUV/AkBwhBwAAAAAJtpv/OFvKJVO6Qf/6gdyKs5GyGGaD15Z5dTlWq6ciiO7ZOvk757Ur/3zX9PNl2+Ou+nYgo4+fVS337zdDDammguPt16hJk+qO/WNZ7G2VtuY1WGtWTp2/thY2z6OQdY4Bhz9ZrzEZTA6aDvDeF1TkD4N61zD6nXtw/Zp0GdmlH0Q9F7E6R4CQDtz3A0AAAAAgGF97dWv6R/8wT9Qbb2m9Z+t6xf/3y+0+rNVrf3NmtZ+tqbyR2XVrbp+4w9/Q7/2z39t3M3FFrZ8ZVlOxVGxUFRppaRivqjih0UV881/lz8qq3KvourPq6p+XFVtrSZrzZIa0tnLZyNtWxSvmel2zris0zBsHe3XF9VaDsP0Y6fyftvZ6bg4DcgH1a8/pdH1ab9yk9CfABB3zOQAAAAAsCV86b/6kr54/ov6T//Xf9KP/92PtfKXK5KkzImMDn/tsL70X35JZoLfeSF6F966oEtPXVLxTnFjdlEilZBMyas/mF1UcTdmcHiep+dvPB9a/f0GVdsHUzcf3+9X7kHq61cmSJ1R17F5tkOnsoP2RbcFxf2U7SVoO/uV62YU961Xnb2293quBxFVn/a754P25zDPGgBsNYQcAAAAALYMM2nq+D88ruP/8Pi4m4JtLHcqp2e/+6xeO/2arHVLM3tmlEglZEwZ8hqeGm5DruVuzOB4/sbzyixFvxZHFIOd3V59tBUGVkd5bcPUFbTsdrx3rX1By2/nPgWAOCPkAAAAAAAgZIuPL+rF2ou69o1reu877ykxk5DXaK50b5iGXMvVsfPHfL+iyu/AbBDDDsAGKT/utQeiOscwdY6j7CivL+g5J+m5HrTcuJ6XsM9JiANg3Ag5AAAAAACIyLnXz+nc6+e08s6K8m/nJTVnemSORz9zAwAAYDsg5AAAAAAAIGKZExllThBsAAAAhI2QAwAAAAAAxN4gC0nz+hwAALYPc9wNAAAAAAAAAAAACIKZHAAAAAAAIPaYnQEAADphJgcAAAAAAAAAAJhIhBwAAAAAAAAAAGAiEXIAAAAAAAAAAICJRMgBAAAAAAAAAAAmEiEHAAAAAAAAAACYSIQcAAAAAAAAAABgIhFyAAAAAAAAAACAiUTIAQAAAAAAAAAAJhIhBwAAAAAAAAAAmEiEHAAAAAAAAAAAYCIRcgAAAAAAAAAAgIlEyAEAAAAAAAAAACYSIQcAAAAAAAAAAJhIhBwAAAAAAAAAAGAiEXIAAAAAAAAAAICJRMgBAAAAAAAAAAAmEiEHAAAAAAAAAACYSIQcAAAAAAAAAABgIiXG3QAAAAAAQHQMw9j47nleoOP8nMMwjED7AAAAgGEwkwMAAAAAtqhWONEKGDaHFX6PawUUrU+nc3Q7b799AAAAwLCYyQEAAAAAW1gruOgWUPQ7rt8MjFYI0i386FfvZoWbBeVv5iVJuZM5ZZeyHY879Kqv033GBy/0L/vBC4Odc/P5upXtVuegdQ3SliD1dCrvp2x7uVHXF1WdYd83P89Kt7oHPb5dkPJR3Iuwn9Eg98LvfRjV3y0AYHiEHAAAAACAQMJ6RdXV5au6de2WkjuTkiHpQTGn4ujo00e1fGU5pBb3d+jV4QbMBzmmtS+MQdNh6+lWvld/BA2b+rU17HYGqTPs++a3r4L26TDC7tNu5Ybp07D6JYz7ki7MtwAAIABJREFUEObfLQAgHIQcAAAAAABfNgcXYQQcd27c0cUnLyq9kNbB0weVmk3JMA01nIbski1r1dLtN2/rlelXdOGtC8qdyknqPxDqZ/Cx1yDsIEGHX6Oor1+/+Bl83rzfb/ta+wYdiO4WLETVzk51tp+337GD1hdUWH3aT9A+DVpu2D4N2i9BjPP+AwAGw5ocAAAAAIC+OgUXhmFsfFr/9rNPagYcl79+WdmlrB7+6sNafHxRuZM5ZY5ntP/z+7X3yF7NPzyvfY/sU/pAWpeeuqQPv/9hpNcY5Jf5fgbVux0T5gCpn4HhQc8xzLV106vfompnv3s1aFvCvOZO5x7HwPmgfRqk3DB9Gka/DBuC+tkHABgPQg4AAAAA2MK6hQyDHNcp4Ni8GPnm9Tz67Wu5+ORF7T28V9mlrHInc3roKw8p++Ws9n9hv/Yc3qP5Q/PatbhLs5lZzWXnlNyR1GunXwvQA9HZCoOdfgOAUb9Gqb2+uLbTrzg+K0H7dJLvRRzvAwBgeLyuCgAAAAC2qNai363gYnPQsDm46Hfc5v+27w/i6vJVpfente+RfVp4bEGZpYzmD82rbtdVvltWYiYhwzDUcBuq23W5liun4shat/TG+Td09ttnh6p/WHEcvEVn414/YdzhEIP6TfzNAsDWRsgBAAAAAFtYt0Ci08yMQcoPclz7vlvXbung6YNKH0hrbnFO84fmtfvgblmrlup2XU7FkV20lZpNNT/plJLppGb2zOjdK+9GFnIMspB4mIuFRz0QzQDvJ8IIAQZdvHqcQUMc2uBHlM9o2H3A3xMAxA8hBwAAAABgZAo3C0ruTCo1m1JyZ1Jm0lTdrstateRarrzGg1klU4bMhCkzacpMmUqkEs3PTEIr76wocyIzVDt6DVQGWbw6aJ2jHHyOw0D3By80+6HTos2jGOjutj1I3wy7tkPYRtmfUYl6Ife4nxMAEAxrcgAAAADAFlQoFD716bX9+9///sb+69evR/r9e69/TzIkwzTkNTw5ZUflu2UV80VV7ldkl2y5lquG25DX8CTvwauyTMmYapbJv50P2i1DiWLQOOqB6Dj/kr8VdrQ+o9BawDroQtZ++3PUAUO/xdPjHHhE+YyGfd1x/nsCgO2MmRwAAAAAsMW8/PLLOnLkiM6cOfOZfZ1eK7V5W9TfmxukhtOQU3JU/biqxExCTsWRYRhyLVe19ZqcsiPXclV36s3Ao+41Q4+QdBuk7DfLYJjBzU5lu9UXhrgOyHZbmDrqBau7BQF+78GgAUcc+r11fXE1ioAj7NdUxeG+AgA+jZADAAAAALag999/X88888xntudyuc9se+KJJza+bw5GIvn+zBn95A9+Irtkq7pa1fS9aRmGIbtoy5hqLjbulB3V1muyy7acyqfDDsM0lDv12WsIS79B714DxkEGQQcZZB/EJAzIxrlt7YL0Z9jPylYThzVpJjG4AgB8FiEHAAAAAGBksktZORVH1qqlyv2KEtMJNdyGUrMpmQlTXsOTa7myy7ZqazXZpQdBR8WVazU/mePDrcex1U3ygGwc2x7HNk26SerTSWorAGxXhBwAAAAAgJE6+vRR3X7zdjPYmGouPJ6aTclMmpIn1Z26nIoju9QMOlqzOqw1S8fOHxtLm3sNcMZpEHTQtviduTLuawvazmFf1xR0dk6Y5xtGr2sftk+DPjOj6oMw7kNcnn8AQG8sPA4AAAAAGKnlK8tyKo6KhaJKKyUV80UVPyyqmG/+u/xRWZV7FVV/XlX146pqazVZa5bUkM5ePhtp26JaqHgU9Q07INvelqjWcuh23qCLevttZ6fjetU5KQPc/fpTGl2f+n3e496n0mS1FQC2O2ZyAAAAAABG7sJbF3TpqUsq3inKqThKppNKpBKSKXl1T3WnLrfibszg8DxPz994PrT6+w3GRrE+RpT1ba6j36/3O21rlelUtt8v8vttH3Rtk26CtrNfuU6G6c9hBO3ToIFUVH3a754P2qfDPGtBjOv+AwCCYSYHAAAAAGDkcqdyeva7z6p0v6TVn642Z3PkiyoVShuzOcr3yioWinJKjp578zlllqJfi+ODF8IduOx3rrDrC6rXYPYo6/LTX4NsD6POuOvV/nH1KQAAo8RMDgAAAADAWCw+vqgXay/q2jeu6b3vvKfETEJew5MkGaYh13J17Pwx36+o8jO4OspB+yjrjKKOQc8xTJ3jKBvWuhrDiOpZGba9ce/TsO9HHP5mAQDhIeQAAAAAAIzVudfP6dzr57Tyzoryb+clNWd6ZI5HP3MDAAAAk42QAwAAAAAQC5kTGWVOEGwAAADAP0IOAAAAANueYRgb3z3PG/i4fuV77fdbN7aXQRaS5tU6AABgO2PhcQAAAADbWitkaAUMm0MHv8d5ntc1oDAMY2O/53mfKtdrHwAAAID+mMkBAAAAYNtrBRT9gga/x0WpcLOg/M0H61aczCm7lB1LOxAtZmcAAAD4Q8gBAAAAABPg6vJV3bp2S8mdScmQ9GDiiFNxdPTpo1q+sjzW9gEAAADjQMgBAAAAABHqNevDz4yQOzfu6OKTF5VeSOvg6YNKzaZkmIYaTkN2yZa1aun2m7f1yvQruvDWBeVO5aK4DAAAACCWCDkAAAAAIGLdFinvt+/OjTu6/PXLyi5lte+RfUofSCu5Mymv4ckpOaquVlW5X1FqNqVioahLT13Ss999NtqLAQAAAGKEkAMAAADAttdaALzfrAq/x/Ur73ffxScvKruUVXYpq4XHFjS3OCczacopO6p+XNX0vWklphMyp0xJUvFOUa+dfk36J4GaBwAAAEwcQg4AAAAA21ortGgFF+0zK9oXG+92XPv31v7N+9pDjF77ri5fVXp/Wvse2aeFxxaUWcpo/tC86nZd5btlJWYSMgxDDbehul2Xa7lyKo6sdUv2VVtiiQ4AAABsA4QcAAAAALa9brMr2rf7PS6Mfbeu3dLB0weVPpDW3OKc5g/Na/fB3bJWLdXtupyKI7toKzWban7SKSXTSc3smZF9y+56XgAAAGArMcfdAAAAAADApxVuFpTcmVRqNqXkzqTMpKm6XZe1asm1XHmNB7NKpgyZCVNm0pSZMpVIJZRIJaSEZKwEe6UWAAAAMEkIOQAAAABsW48++qgKhcLGp2Xzths3bmxsv379+ki+52/mJUMyTKO5yHjZUfluWcV8UZX7FdklW67lquE25DU8yXvw6itTzeDDMKV84G4BAAAAJgavqwIAAACwLb300kvK5/MdXxm1edu4vsuTGk5DTqm5yHhiJiGn4sgwDLmWq9p6TU7ZkWu5qjv1ZuBR95qhBwAAALBNEHIAAAAA2LZyuVzf7Zu/nzlzZiTfcyebddolW9XVqqbvTcswDNlFW8ZUc7Fxp+yotl6TXbblVNrCDs+TlyPsAAAAwNZHyAEAAAAAMZNdysqpOLJWLVXuV5SYTqjhNpSaTclMmPIanlzLlV22VVuryS49CDoqbnPNDteTMuO+CgAAACB6hBwAAAAAEENHnz6q22/ebgYbU82Fx1OzKZlJU/KkulOXU3Fkl5pBR2tWh7VmSUfH3XoAAABgNAg5AAAAACCGlq8s65XpV1QsFCVJruUqlU7JTJkyjOYrq1zLlVNpvraqtlZrBhwNScuSeFsVAAAAtgFz3A0AAAAAAHR24a0LckqOineKKuaLWs+vq5QvqVgoqrRSUvleWZV7FVU/rqrycUWe5+n5G8+Pu9kAAADAyBByAAAAAEBM5U7l9Ox3n1XpfkmrP11VMd8MO0qFUjPk+Kis8r2yioWinJKj5958TpklFuMAAADA9sHrqgAAAAAgxhYfX9SLtRd17RvX9N533lNiJiGv0XwXlWEaci1Xx84f09nLZ8fcUgAAAGD0CDkAAAAAYAKce/2czr1+TivvrCj/dl5Sc6ZH5jgzNwAAALB9EXIAAAAAwATJnMgoc4JgAwAAAJAIOQAAAABgSzMMY+O753mBjuu2b/P29v299gEAAABhIeQAAAAAgC2qFTR4nifDMGQYRsegoddx7WU2/9tvGNK+DwAAAAgLIQcAAAAAbGGbA4leQUO34/zOvugWoPTbt1nhZkH5mw/WGzmZU3Yp2/G4Q6/6atJnfPBC/7IfvNB9X7eyvcr0Ku+3nF9ht89v2aDXFVZ9UdU5bH/2Ol+/cwzap8M8193KR3EvRv03FMY9HOZ5AwCMBiEHAAAAAKCnIKHHIK4uX9Wta7eU3JmUDEkPqnAqjo4+fVTLV5ZDr7ObQ692HsDsNYjc2jfoYHmYhmlfr/Ld+qNfnb30a2vY7QxS57D9OUj9QY4LU9h9Gpe/oTDuYdC+AQCMFiEHAAAAAKCnbq+r6rXNz747N+7o4pMXlV5I6+Dpg0rNpmSYhhpOQ3bJlrVq6fabt/XK9Cu68NYF5U7lJPUfCPUz+NhrELbbAGa/evsNfLb2RTWQHbR93WYYRH1d3YKFqNrZqc728/Y7dtD6ggqrT/sJ2qdBy436b2iY+sJ43gAAo2GOuwEAAAAAgO3nzo07uvz1y8ouZfXwVx/W4uOLyp3MKXM8o/2f36+9R/Zq/uF57Xtkn9IH0rr01CV9+P0PI22Tn8HVQfe19kc9GDpM+7odF8V19Qqiompnv/Br0LaEec2dzj2OgfNB+zRIuVH/DYV1D4P2DQBgdAg5AAAAAGALa71qqt/C392Oi2rB8ItPXtTew3uVXcoqdzKnh77ykLJfzmr/F/Zrz+E9mj80r12LuzSbmdVcdk7JHUm9dvq1SNqynfkNAEb9GqX2+uLaTr/iODAetE8n/V74sR2uEQC2El5XBQAAAABbVGsR8VZQ0e21U72O67cQeZBXVV1dvqr0/rT2PbJPC48tKLOU0fyhedXtusp3y0rMJGQYhhpuQ3W7Ltdy5VQcWeuW3jj/hs5++2zAHsF2E2T9jCjqH1d9cQxXAAAIGyEHAAAAAGxh3QKI9u29FhQPe9+ta7d08PRBpQ+kNbc4p/lD89p9cLesVUt1uy6n4sgu2krNppqfdErJdFIze2b07pV3Iws5olhIOw7i3r5RCiMEGHQR8XEGDXFogx/jDoMAAJONkAMAAAAAMDKFmwUldyaVmk0puTMpM2mqbtdlrVpyLVde48GskilDZsKUmTRlpkwlUonmZyahlXdWlDmRGaodvQY5gw4Ix30gOQ7t++CFZt93WrQ5yoHnbuceJgQIusB9VEbZn1EZ9TMah78JAMDwWJMDAAAAALagRx99VIVCYePTsnlbv+3vvPPOxv7r16+H8v17r39PMiTDNOQ1PDllR+W7ZRXzRVXuV2SXbLmWq4bbkNfwJO/BuiCmZEw1y+Tfzg/ZO+GJ+y/l49y+VtjR+oxCawHroAt8++3PUQcM/RZPj3PgMepnNM5/EwCAYJjJAQAAEFO93oHv57h+5fu9Y99P3QDi6aWXXlI+n+/49+v39VXt28L63twgNZyGnJKj6sdVJWYSciqODMOQa7mqrdfklB25lqu6U28GHnWvGXqEpNsAZ7dZBt2O7XWucYtr+7ot2hz1Ys7dggC/93zQgCMO/d66vrgi4AAAhIGQAwAAIIY2L/7bWgy4V1DR6bjWfzcHFpvLdVuAuNc+AJMjl8sNvX1paWnj+5kzZ8L5/swZ/eQPfiK7ZKu6WtX0vWkZhiG7aMuYai427pQd1dZrssu2nMqnww7DNJQ71fkawuB30Dvug6Vxb58U77a1C9KfvcKFSbg/USPgAACEhZADAAAgpjaHFZ2CikGP61RmWA23oVtXbumv/++/VuEvmq+3yZ7I6sjXjujobx2VMeWvPQC2j+xSVk7FkbVqqXK/osR0Qg23odRsSmbClNfw5Fqu7LKt2lpNdulB0FFx5VrNT+b4cOtxDCvug6Vxb18vcWx7HNs06Qg4AABhYk0OAAAABPLD7/xQ/2z/P9Mf/84f68ff/bGcoiN7zdb7f/y+/ui3/0j/dOGf6kfXfjTuZgKIoaNPH22uwXGvotJKScV8sfkpFFVaKal8r6zK/Yqqv6iqtlbbmNVhrVk6dv7YWNse98HSQdvX7xVRcbneoO0ctt1Brr993Y9Oa4AEXRNkUL1mkwzbp0GfmUkIOCbl7wIA0ETIAQAAsM21v45q86uvus0M+Q//w3/QH3/zj7XwyIIe/nsP65ef+mUdPH1Qi08savGJRX3uC5/Tjj079G+e+zf60//pT0d1KQAmxPKVZTkVZyPUKOaLKn7YDDpKKyWVPyqrcq+i6s+rqn7cDDqsNUtqSGcvn420bZP8iqFh29d+7VGt5TDswHHQdnY6rledcb/fLf36Uxpdn/YrNwkBR7dzdPs3AGD8eF0VAADANtZtvY1uC5hL0p/8zp/ove+8p1/6z39Jew/v1Vx2TokdCTWchmrrNVU/rqp8r6zEjoTMlKk//9//XLW1mrQQ+eUAmCAX3rqgS09dUvFOUU7FUTKdVCKVkEzJq3uqO3W5FXdjBofneXr+xvOh1d9voLJ9UHTz8X5+He+nrm4LbwcxTPs2L07dqWy/X+T32z5In/QStJ39ynUyTH8OI2ifBh14j6pP+93zUfwNDXsPg/YNAGD0mMkBAAAQU61wod86G36P61Su39oc7ces/MWK/vL//EvlTuV04EsHlDuV0+ITi8ocz2jfo/s0//C8dj20S3PZOaU/l1Z6f1qzmVn94A9/IN0dqHkAtrjcqZye/e6zKt0vafWnqxuvrCoVShuzOcr3yioWinJKjp578zlllqJfi2NUrxGKm14DvaOsq199Qds5TJ1x16v94+rTrWI7XCMAbAXM5AAAAIihza+Mav27ZXPw0O+49u+t/Z2CkfZ97eeTpH/72/9W8780r/n/bF57D+/VgS8d0N5f3qvaek2lQkmGaajhNuTWXDlVR07F2Vg82P0jVwrvR9gAtoDFxxf1Yu1FXfvGNb33nfeUmEnIazz4f5FpyLVcHTt/zPcrqvwMPAYdnBxmUHMUA6Jh1DHoOcbVJ6O4h1Hds6DBwbDnjar8qPo0SNmw7iGBBgDEHyEHAABATHWbZdG+3e9xw+6zfmGp8E5Bh04f0s59O5ufvTuVmk2p4TSU2JlQckey4yc1m1LlbypSrWu1ALaxc6+f07nXz2nlnRXl385Las70yByPfuYGAAAAJhshBwAAAHzJ38xrKjWlxI6EplJT8jxvYwZH3amrbtebv8A2mr/ANqYMmQmz+UmaMhKGlB/3VQCIs8yJjDInCDYAAADgH2tyAAAAxMgXv/hFfe1rX9MPf/jDjU/L7//+74/1+71b92ROmZIn1e26asWaSh+VVMwXVb5XVm2tJqfiqF6rq+E25DW8T16BZRqaMqbk3e29BggAoOnQq/4/AAAA2xkzOQAAAGLk3Llzsiyr477f+q3fGuv3udxcc70Ny1Vtvabq/aqmElOyy7ZM05Rbc2WXbNklW071QdjhNNSoN+TVPdW9uoxdgy2ODgAAAABAL4QcAAAAMTMzM9Nx+8LCwli/576ck9fwVCvWVP1FVam7KXmep1QxJTNhNgOQqqtasSa7aMspOxthR91pvsrKyzGTAwD8YLFjAAAAfwg5AAAA4Mvug7uV/lxa1s8tVXZXNJWcUt2pKzWb0lRySl7Dk1tz5ZQd1dZrqhVrzaCj0vxoVtKucV8FAAAAAGArIeQAAACAL8aUob/13/0t/en/+KdKziZlmIbqdl3JdFJTySlJUt2py602X1tVW6+ptl5rvr6q4kh/VxJvqwIAAAAAhIiQAwAAAL498Y+f0Dv/6h2t/mxVnufJtdyNkMMwDDXqjeZsjooju9gMOir3K1p4bEGFUwWJt1UBAAAAAEJkjrsBAAAAmCy//YPfVnp/Wqs/WVXxw2Lzky+quFJUaaWk8t2yKvcrqn5cVemjknb/0m598//95ribDQAAAADYgpjJAQAAgIEYU4b+0fv/SJf/i8u68//cUfluWYkdCSWmE/I8T/VaXXbF1lRySr/81V/W+T86P+4mAwAAAAC2KEIOAAAABPLMnzyjv/53f60b/+sNffQXH6n0i5IkKbUrpYe+8pD+9n//t3X4qcNjbiUAAAAAYCsj5AAAAEBgR379iI78+hFJUm29Jkma3jU9ziYBAAAAALYRQg4AAACEgnADAAAAADBqLDwOAAAAAAAAAAAmEiEHAAAAAAAAAACYSIQcAAAAAAAAAABgIhFyAAAAAAAAAACAiUTIAQAAAAAAAAAAJhIhBwAAAAAAAAAAmEiEHAAAAAAAAAAAYCIRcgAAAAAAAAAAgIlEyAEAAAAAAAAAACYSIQcAAAAAAAAAAJhIiXE3AAAAANhKDMPY+O55XqDjgu4bpH4AAAAA2AqYyQEAAACEpBUwtMKFzYGD3+MMw5DneRuf9n29zt9eFgAAAAC2OmZyAAAAACFqhQvtAYXf4/qFE93KtQKOQRRuFpS/mZck5U7mlF3Kdjzu0KsDnXbDBy/0L/vBC4Odc/P5+pXtVPeg9Q16/kHqCNq+9nKjri+qOoftz17nG/RZCfJsDVs+insR9jMa5F74uQ9+/x8T5t8vAADYOgg5AAAAgJgJ+sopv+WuLl/VrWu3lNyZlAxJDw51Ko6OPn1Uy1eWB25zUIdeHW7AfNDjWvuGHSwdto5u5Xv1R9CwqV9bw25nkDrDvmdhPCtRCbtPu5Ubpk/D6pdx9C8AANh+CDkAAACAmNkcUAwyQ6NfuTs37ujikxeVXkjr4OmDSs2mZJiGGk5DdsmWtWrp9pu39cr0K7rw1gXlTuUk9R8I9TP43GsQdpCgo59+vxoPs85+/eJn8Hnzfr9ta+0bdAC5W7AQVTs71dl+3n7HDlpfUGH1aT9B+zRouWH7NGi/DMpPuMMsDgAA0A1rcgAAAADbwJ0bd3T565eVXcrq4a8+rMXHF5U7mVPmeEb7P79fe4/s1fzD89r3yD6lD6R16alL+vD7H0bapiC/zPdbpttxYQ2U+hkYHvQcfgKDoIPqgwyA9zvO76uYBrkHYfSn3/rbzz2OwfNB+zRIuWH6NIx+IZwAAACjQsgBAAAAhKj1yqhe63H0Oi5ouX4uPnlRew/vVXYpq9zJnB76ykPKfjmr/V/Yrz2H92j+0Lx2Le7SbGZWc9k5JXck9drp1waqI2pbYbDUbwAw6tf8tNcX13b6FcdnJWifTvK9iPK1cAAAAC28rgoAAAAISWsx8FYA0e31Ub2O67UQedByV5evKr0/rX2P7NPCYwvKLGU0f2hedbuu8t2yEjMJGYahhttQ3a7LtVw5FUfWuqU3zr+hs98+G0r/BMVA5+QY96/3xx0OxTFcGYew7wP9CgAAemEmBwAAABAiz/M2Pu3b/RwXxb5b125pz+E9Sh9Ia25xTvOH5rX74G7tXNipmfkZTe+aVmo29cknnVIyndTMnhm9e+XdIN3gi5+B0KCvqeq1SPMg5wuCUOYTh1799CfoOQY5bpwD4sNc5yhF2cY43AcAALC9MJMDAAAA2MIKNwtK7kwqNZtScmdSZtJU3a7LWrXkWq68xoPZIVOGzIQpM2nKTJlKpBLNz0xCK++sKHMiM1Q7eg2qBlm8ut/xvQabRzX4GodB3s190W3h8ShEETIFXeA+KqPsz6hEvZD7MAhLAACAX8zkAAAAAELw6KOPqlAobHxaNm/rtP3P/uzPNrZdv3499O/fe/17kiEZpiGv4ckpOyrfLauYL6pyvyK7ZMu1XDXchryGJ3kP1vswJWOqWSb/dn6IngluEgeN4zwwG8asikG1FrAOupC13/4c9bPSb/H0OD+7UT6jcb5uAACwdTGTAwAAABjSSy+9pHw+3/UVUp1sXp+j07FhfW9ukBpOQ07JUfXjqhIzCTkVR4ZhyLVc1dZrcsqOXMtV3ak3A4+61ww9QtJtQLXfLIOwB8a71ReGuAYc3V7hFfWC1d2CAL/3YNCAIw793rq+uBpFwBGH+wAAALYXQg4AAAAgBLlcLtD2zfvPnDkT/vdnzugnf/AT2SVb1dWqpu9NyzAM2UVbxlRzsXGn7Ki2XpNdtuVUPh12GKah3KnO1xCGfoPevQaMOw2q9htoHWSQfRCTMMAb57a1C9Kfgz4r282o+iCM+8D9AgAAgyDkAAAAALaw7FJWTsWRtWqpcr+ixHRCDbeh1GxKZsKU1/DkWq7ssq3aWk126UHQUXHlWs1P5vhw63FsdZM8IBvHtsexTZOOPgUAAFsZIQcAAACwxR19+qhuv3m7GWxMNRceT82mZCZNyZPqTl1OxZFdagYdrVkd1pqlY+ePjaXNvQZj4zRgO2hb/M5cGfe1BW3nsK9rCnL9cXpWel37sH0a9JkZVR+EdR/i/LovAAAQTyw8DgAAAGxxy1eW5VQcFQtFlVZKKuaLKn5YVDHf/Hf5o7Iq9yqq/ryq6sdV1dZqstYsqSGdvXw20rZFNaDZ7bxh1jfs4HF7W8bVF4Mu6u23nZ2O67deip/2jJufZ2tUfdqv3KT0aSeT2GYAADAezOQAAAAAtoELb13QpacuqXinKKfiKJlOKpFKSKbk1T3Vnbrcirsxg8PzPD1/4/nQ6u83GBvWgObmmQT9flU/jM3nHrSefm3s94v8ftsHXdukm6Dt9HsPurUvyvvWq95e29vrDRpIRdWn/e75oH06zLMGAAAwaszkAAAAALaB3Kmcnv3usyrdL2n1p6vN2Rz5okqF0sZsjvK9soqFopySo+fefE6ZpejX4vjghfAHSnudM4r6gujVvlHW1a++oO2Me/8Po1f7x9WnW8UkzzwBAADjw0wOAAAAYJtYfHxRL9Ze1LVvXNN733lPiZmEvIYnSTJMQ67l6tj5Y75fUeVnIHKUg/ajqDvMcw96jmHqHEfZsNbVGEbQ4GDY80ZVflR9GkXwOI56AQDA9kDIAQAAAGwz514/p3Ovn9PKOyvKv52X1JzpkTke/cwNAAAAAAgTIQcAAACwTWVOZJQ5QbABAAC+yd+bAAAgAElEQVQAYHIRcgAAAADY9gZZSJpX6gAAAADxwcLjAAAAAAAAAABgIjGTAwAAAMC2x+wMAAAAYDIxkwMAAAAAAAAAAEwkQg4AAAAAAAAAADCRCDkAAAAAAAAAAMBEIuQAAAAAAAAAAAATiZADAAAAAAAAAABMJEIOAAAAAAAAAAAwkQg5AAAAAAAAAADARCLkAAAAAAAAAAAAE4mQAwAAAAAAAAAATCRCDgAAAAAAAAAAMJEIOQAAAAAAAAAAwEQi5AAAAAAAAAAAABOJkAMAAAAAAAAAAEwkQg4AAAAAAAAAADCRCDkAAAAAAAAAAMBEIuQAAAAAAAAAAAATiZADAAAAAAAAAABMJEIOAAAAAAAAAAAwkRLjbgAAAACwlRiGsfHd87yhjjMM41P7NpdpL9e+r1/9AAAAALAVMJMDAAAACEkraGiFC52CB7/HdSvred7Gp9t2wg0AAAAA2wUzOQAAAIAQtQIGz/O6BhX9jmvN4OhVvpf2GSDdFG4WlL+ZlyTlTuaUXcp2PO7Qq4GaoQ9e6F/2gxf6n6fTOaIs51e3a/NbR1jXNer6oqpz2P7sdb5+5xi0T4d9rkd1L8J+RqN+1gAAAIIg5AAAAABipF9A4fd1WL1cXb6qW9duKbkzKRmSHpzGqTg6+vRRLV9ZDnTeIA69GmyQtle5XgPQ/erzq18d0mivq5dh+iNIO4PUOWx/DlJ/kOPCFHafdis3TJ9G9az1qhMAACAoQg4AAAAgJvoFHO37Oh3f6xx3btzRxScvKr2Q1sHTB5WaTckwDTWchuySLWvV0u03b+uV6Vd04a0Lyp3KSeo/EOpn0LLXIKyfQdrN+/uV61an33J+9euXqK9r0IHoQftj2HZ2qrP9vP2OHbS+oMLq036C9mnQcsP26SD90m/mzCjuIwAA2J5YkwMAAACIEcMwNj6tf4fhzo07uvz1y8ouZfXwVx/W4uOLyp3MKXM8o/2f36+9R/Zq/uF57Xtkn9IH0rr01CV9+P0PQ6m7G78Dne3H+fm1+yADvkEMEq74Pc5PYBB0UH2Y/hi0nf3Cr0HbEuY1dzr3OAbcB+3TIOWG6dNh+mWQ+w4AABAGQg4AAAAgRH7DiU7HdVo8PKxFxC8+eVF7D+9Vdimr3MmcHvrKQ8p+Oav9X9ivPYf3aP7QvHYt7tJsZlZz2TkldyT12unXQqk7CL8D5VG8VidKUV9XUO31xbWdfsVxQD1on076vQAAAIgar6sCAAAAQtJaLLwVXGwOKDa/RqrXcb20Byd+X1V1dfmq0vvT2vfIPi08tqDMUkbzh+ZVt+sq3y0rMZOQYRhquA3V7bpcy5VTcWStW3rj/Bs6++2z/jsB29q4110YdzgUx3AFAABgq2MmBwAAABCi9pkYm7f7Oc5vmU7lup3r1rVb2nN4j9IH0ppbnNP8oXntPrhbOxd2amZ+RtO7ppWaTX3ySaeUTCc1s2dG71551++lD2yr/vJ8q15XEIde/fQn6DkGOW6cQcMw1zlKUbSx34ySONwfAACwNTGTAwAAANjCCjcLSu5MKjWbUnJnUmbSVN2uy1q15FquvMaDWSVThsyEKTNpykyZSqQSzc9MQivvrChzIjNUO3oNqoa9VkZroLnbQtujEofB3HH1RxQD3UEXuI/KuJ+vMITdX5uft1HUBwAAIBFyAAAAAKEoFAqf+nc2mx1q+9TUlH70ox/pySeflCRdv3490Pf8zbxkSIZpyGt4csqOynfLqtt1eQ1PdsmWa7lquA15DU/yHrwWy5SMqWaZ/Nv5oUOOcRjHoHOcf60+jv4YNgjw259xCLBaA/yt9sTxGZDi/YwCAAAEQcgBAAAADOnll1/WkSNHdObMmc/s6/YKKT/bw/ouT2o4DTklR9WPq0rMJORUHBmGIddyVVuvySk7ci1XdafeDDzqXjP0CEm3AdVuswzCqKvbeglRDYjHdfB4XP3RKwjwc88HDTji0O+bg444irqvep0/ir91AAAAiZADAAAACMX777+vZ5555jPbc7lcx+P9bP/c5z638X1zgDLI99zJ5vnskq3qalXT96ZlGIbsoi1jqrnYuFN2VFuvyS7bciqfDjsM01DuVOe2hmGQQe8g5x6VOA20dxPntrUL0p+9woVJuD9RG2fA0dpO0AEAAKJAyAEAAABsYdmlrJyKI2vVUuV+RYnphBpuQ6nZlMyEKa/hybVc2WVbtbWa7NKDoKPiyrWan8zxyXtVVTdRDPRO8gB6HNsexzZNOvoUAABsZea4GwAAAAAgWkefPqpivqjKvYpKKyUV88Xmp1BUaaWk8r2yKvcrqv6iqtpabWNWh7Vm6dj5Y2Npc79XKcVl0HbQdkzKdQVt57DtDnL9H7zQ/dN+TNR6zSYZtk+DPjNxeaYAAACiQsgBAAAAbHHLV5blVJyNUKOYL6r4YTPoKK2UVP6orMq9iqo/r6r6cTPosNYsqSGdvXw20rb5Wb+g/Zh+ZUYVIAx7vkGvK6hh+yNoOzsd12/NBj/tGbd+/SmNrk/9/i2M49VtfrcDAAAMi9dVAQAAANvAhbcu6NJTl1S8U5RTcZRMJ5VIJSRT8uqe6k5dbsXdmMHheZ6ev/F8aPX3G+DstUh1t/K9Bm6jHlDdfH4/v95v3xbkuvwOHvcKEAYRtJ39ynUyTH8OI2ifBn2+ourTfvd80D4N0i9+73vcQywAADB5mMkBAAAAbAO5Uzk9+91nVbpf0upPVzdeWVUqlDZmc5TvlVUsFOWUHD335nPKLEW/Fke/1wj1GvQNUiYuA6xBriuKuvrVF7Sdk3APgur37I2jT+Oi1/VvhXsPAADiiZkcAAAAwDax+PiiXqy9qGvfuKb3vvOeEjMJeQ1PkmSYhlzL1bHzx3y/osrPgGUYg5pBzhH1YOo4rmuYOsdRdtB1NaIQNDgY9rxRlR9Vn47r+gAAAIIg5AAAAAC2mXOvn9O5189p5Z0V5d/OS2rO9Mgcj37mBgAAAACEiZADAAAA2KYyJzLKnCDYAAAAADC5CDkAAAAAbHuDLCTNq3gAAACA+GDhcQAAAAAAAAAAMJGYyQEAAABg22N2BgAAADCZmMkBAAAAAAAAAAAmEiEHAAAAAAAAAACYSIQcAAAAAAAAAABgIhFyAAAAAAAAAACAiUTIAQAAAAAAAAAAJhIhBwAAAAAAAAAAmEiEHAAAAAAAAAAAYCIRcgAAAAAAAAAAgIlEyAEAAAAAAAAAACYSIQcAAAAAAAAAAJhIhBwAAAAAAAAAAGAiEXIAAAAAAAAAAICJRMgBAAAAAAAAAAAmEiEHAAAAAAAAAACYSIQcAAAAAAAAAABgIhFyAAAAAAAAAACAiUTIAQAAAAAAAAAAJhIhBwAAAAAAAAAAmEiJcTcAAAAAAOLMMIyN757nBTqu2z6/ZfzUDwAAAGxHzOQAAAAAgC5aQUMrXOgUPPQ7zjAMeZ638WkPNroFF5vLEG4AAAAAnTGTAwAAAAB6aAUM7QGF3+PCCChaQUk/hZsF5W/mJUm5kzlll7Idjzv0arB2fPBC/7IfvNB9X7eyvcr0Kx+0rN9yQc49aB1BryvoNYVVX5R1Dtun3c7Vr/ygfTrM30O38lHci7Cf0UHvQ5DrjPJvFgCwtRByAAAAAMCEu7p8Vbeu3VJyZ1IyJD3IQ5yKo6NPH9XyleWRteXQq50HI3sNBrf2dRvEjKpsGIZpW6/y3fqxX5299Gtr2O0MWuewfer3XEGOC1PYfTrqvz0/wr7/AAB0QsgBAAAAACPid0aG3zJ3btzRxScvKr2Q1sHTB5WaTckwDTWchuySLWvV0u03b+uV6Vd04a0Lyp3KSeo/oOlnALnX4GS3wct+9XYq1+9X+P3qbC8bxQBqkOtqb8vm/VFfU7c+jqqdnepsP6+f4wetM4iw+rSfoH0atNyw/Tns30+QMCPKv1kAwNbCmhwAAAAAMAJBAo5e7ty4o8tfv6zsUlYPf/VhLT6+qNzJnDLHM9r/+f3ae2Sv5h+e175H9il9IK1LT13Sh9//MLT6O/EzSDrovn7H+Blcj/I1N8NeV6fjorimXgFWVO3sF5oFuaeDXPegod04Xoc0aJ8GKTdMfw7TL8Pcf15NBQAYBCEHAAAAAPTQWl+j13oc/Y4LO+CQpItPXtTew3uVXcoqdzKnh77ykLJfzmr/F/Zrz+E9mj80r12LuzSbmdVcdk7JHUm9dvq1UNuA4PwOAI/6V+zt9cW1nX7FcbA8aJ9O+r0AACAqvK4KAAAAALpoLSLeCi42BxWbg4t+x23+7+b9m7f1q2Ozq8tXld6f1r5H9mnhsQVlljKaPzSvul1X+W5ZiZmEDMNQw22obtflWq6ciiNr3dIb59/Q2W+fHa5jgD4GXTsjqvrHVV8cw5VRGvf9BwBsL4QcAAAAANBDtxkY7dv9Hud3X6/9t67d0sHTB5U+kNbc4pzmD81r98HdslYt1e26nIoju2grNZtqftIpJdNJzeyZ0btX3o0s5IhiQewPXmju77dGQRwHU/lF/SfCCgH89Gkcnok4tMGPUT2jhEAAgCgRcgAAAADABCncLCi5M6nUbErJnUmZSVN1uy5r1ZJrufIaD2aVTBkyE6bMpCkzZSqRSjQ/MwmtvLOizInMUO0Ismh0P71ew9MKOsKsb1Ti0L5eYVGUA93dzj1sCBBkofOojLI/oxJVf0V1/wEA2Iw1OQAAAACgg0Kh8KlPp+0fffSRrl+/vrFvFN/zN/OSIRmmIa/hySk7Kt8tq5gvqnK/Irtky7VcNdyGvIYneQ9ehWVKxlSzTP7t/DBdE6qtOtgZ5+tqhR29gqOwtRaTjnIh683HjEq/hdzjHHiM8hkN4/4DANANMzkAAAAAoM3LL7+sI0eO6MyZM5/Z1+s1VSP77kkNpyGn5Kj6cVWJmYSciiPDMORarmrrNTllR67lqu7Um4FH3WuGHiHpNlDZ69VSnY7tdS4/xw1S36jENeDotjB11AtWdwsCwn5W4tTvreuLq1EHHJ22xfFvFwAwmQg5AAAAAKCD999/X88888xntudyuU/9+8CBAxvfN4ciUX0v7GrOKrFLtqqrVU3fm5ZhGLKLtoyp5mLjTtlRbb0mu2zLqXw67DBMQ7lTn76GMPkdvAwj4BikvlGJ00B7N3FuWyeD9mmvcGES7k/U6AMAwFZDyAEAAAAAEyS7lJVTcWStWqrcrygxnVDDbSg1m5KZMOU1PLmWK7tsq7ZWk116EHRUXLlW85M5Ptx6HMPaqoOsk3xdcW17XNs1qehPAMBWxJocAAAAADBhjj59tLkGx72KSislFfPF5qdQVGmlpPK9sir3K6r+oqraWm1jVoe1ZunY+WNjbftWHWQd9Lr6vSIqLv0UtJ1htDtIn3b7tB8TtV6zSYbt06DPzLhfUQUAQFQIOQAAAABgwixfWZZTcTZCjWK+qOKHzaCjtFJS+aOyKvcqqv68qurHzaDDWrOkhnT28tlI2xbVq4L6DeyO07CDx+3XENU1DRuoBG1np+PiNCAflJ9nclR92q/cOPszyP0HAGAQvK4KAAAAACbQhbcu6NJTl1S8U5RTcZRMJ5VIJSRT8uqe6k5dbsXdmMHheZ6ev/F8aPX3G1RtH7zcfLyfX7lv/nfr+EHK9aur2+Lbgwp6Xa1tva4timsKEqAEbaffe9dumD4NKmifBg2kourTfvd80P4c5lkL4/4PWicAYHtiJgcAAAAATKDcqZye/e6zKt0vafWnqxuvrCoVShuzOcr3yioWinJKjp578zlllqJfiyOK1wH1OueoXj8UlV7XNcq6+tUXtJ1b9b5Jvds/rj6Nm618/wEA8cFMDgAAAACYUIuPL+rF2ou69o1reu877ykxk5DX8CRJhmnItVwdO3/M9yuq/Aw6Bh2YDGNAM8g5oh5IHcd1DVPnOMqO8vqCnnNcz/Uo6h3X8xL0HIQfAIBBEXIAAAAAwIQ79/o5nXv9nFbeWVH+7byk5kyPzPHoZ24AAAAA40TIAQAAAABbROZERpkTBBsAAADYPgg5AAAAAAzNMIyN757nBTqu3zm67fdbN+DHIAsk81odAACA8WPhcQAAAABDaYUMrYBhc+jg9zjDMOR53san/Rzt+/2WAwAAALC1MZMDAAAAwNBawUO/oKHbcf1mf3TbH2TmRuFmQfmbD9atOJlTdik78DmwdTE7AwAAYLIQcgAAAACIvTBeSXV1+apuXbul5M6kZEh6cBqn4ujo00e1fGU5hJYCAAAAGCVCDgAAAACx0mnmRqdXVPkpJ0l3btzRxScvKr2Q1sHTB5WaTckwDTWchuySLWvV0u03b+uV6Vd04a0Lyp3KhX9RAAAAACJByAEAAAAgNnq9mipIuTs37ujy1y8ru5TVvkf2KX0greTOpLyGJ6fkqLpaVeV+RanZlIqFoi49dUnPfvfZMC4FAAAAwAgQcgAAAAAYWitk6Lfwd6/jwg44JOnikxeVXcoqu5TVwmMLmluck5k05ZQdVT+uavretBLTCZlTpiSpeKeo106/Jv2TgZsBAAAAYAwIOQAAAAAMpRVatIKLbq+W6nfc5v9u3t9rkfJe5a4uX1V6f1r7HtmnhccWlFnKaP7QvOp2XeW7ZSVmEjIMQw23obpdl2u5ciqOrHVL9lVbYokOAAAAIPYIOQAAADByfheR7jew3a28n/MHnTWAzrr1Za+1NfxsH6bcrWu3dPD0QaUPpDW3OKf5Q/PafXC3rFVLdbsup+LILtpKzaaan3RKyXRSM3tmZN+ye7YHAAAAQDyY424AAAAAtpf2X/F3e71Rr+M8z+sbXrQ+3V6LhK2tcLOg5M6kUrMpJXcmZSZN1e26rFVLruXKazyYVTJlyEyYMpOmzJSpRCqhRCohJSRjhecEAAAAiDtCDgAAAIzc5tcQhXFcpzLdBJnBUfhBQYUfFAYqs10UCoVPffptv379+ki+52/mJUMyTKO5yHjZUfluWcV8UZX7FdklW67lquE25DU8yXsQfplqBh+GKeUDdwsAAACAEeF1VQAAANg2Bgk4/uP/9h/15//Hn2v1g1VNz01LkmrFmuYPzeuJ/+YJfeWFr0TZ1Inw8ssv68iRIzpz5sxn9vl5vVTU3+VJDachp9RcZDwxk5BTcWQYhlzLVW29JqfsyLVc1Z16M/Coe83QAwAAAMBEIOQAAADAlrU51PAbcFhrlv7lyX8p6xeWdi7s1KG/c0jGlNEcLK84Kt0t6XsvfU83/+Cmvvn2N5WaTUV9GbH2/vvv65lnnvnM9lwu1/H4zYFIlN9zJ5v12yVb1dWqpu9NyzAM2UW7eT/dhpyyo9p6TXbZllNpCzs8T16OsAMAAACIO0IOAAAAbEmdQo32tTjaj6n+vKpvffFbSqVTOvh3Dmr2wKyS6WTzdUcVR7W1mnbs26HKvYrW/2Zd/+KRf6HfvfW7I7keDCa7lJVTcWStWqrcrygxnVDDbSg1m5KZMOU1PLmWK7tsq7ZWk116EHRU3OaaHa4nZcZ9FQAAAAD6IeQAAADAyLXChX4LgPs9rlu5zToFHu3bvvXFb2lm94wWv7KohUcXNJudVWI6Iaf6YLD8XkWJHQlNJadkGIbWframb33xW9I3B2oeRuTo00d1+83bzWBjqrnweGo2JTNpSp5Ud+pyKo7sUjPoaM3qsNYs6ei4Ww8AAADAD0IOAAAAjFQrtGgFF5uDhs3BQ7/j2r9vfi1V+zF+XlP17//bf6+6XdfnvvQ57f/CfmV+JaPdB3erUW+o+vOqSh+VZE6Z8jxPDbeheq0u13K1+sGq9KakrwbpDURp+cqyXpl+RcVCUZLkWq5S6ZTMlCnDaL6yyrXc5iyd9Zpqa7VmwNGQtCyJt1UBAAAAsWeOuwEAAADYfjzP2/i0b/d7XPvHz75udXl1Tz/4wx9o75G92rW4S7sP7tbeR/Zq4fMLmsvNace+HZqZn9H0rmlNzzU/qbmUUrP/P3v3HyVXed95/nNv/eiWultqqdWoqlqWkCNksCwsNRJxghkU2zFOYntiJDYYzhzWIrZPwmbIOsmc2c2yhLPsnOzZzeZ4PI5n/QPNibUQUMRsJvGv4BiTQ5g1ILANK2xsBWSL6pZaEt1dVbdu3Z/7R6maVtP1+3f3+3XOPaq+9z73ee5zb7fx91vP88Q1ODooPScC4j3q8FOH5WZdZc5klElnNJ+eVzadVWYqo+x0VrmZnKwZS/mLeVkXLYVhqLuevqvbzQYAAABQI5IcAAAAWPUu/OSCAi/Q4Oig4iNxxdbEJEluzlXgBzJMQ2bUlBkzFYlH3twGIooORmUEhnSxyzeBZaX2p3THN+9Q9nxWs6/NKpMuJjuyU9likuNsTrmZnDJTGblZV3c+fqcSkyzGAQAAAPQLkhwAAADomM2bN2tubm5hK/nJT37S1c9TJ6YkQ4rEIlJYnNYofz6vTDqj/MW8nJwjv+Ar9C+NDDEkwzRkRIrJD8MwZKTrWzcEnTNx/YTuLdyrnR/eqfkz85pPz2v29Kzmfj6n7NmssmezuuY3r9Ef239MggMAAADoM6zJAQAAgI741Kc+pQsXLmhmZmZh3/r16yVJzz77rK666qqufbbfsGWYxsJC1Pk38opNx+RarmRIXt5TIVOQm3flF3wFbqDADxQGxaSHKVOBHbSv89ASBx8+qIMPH9T0C9NKP5eWVBzpkdhDYgMAAADoVyQ5AAAA0BHJZFLJZHLZY7fffntXP29+92aFQSjXcmXP2bJmLBmmoUK2INM0i8mPXHFxaifnyMt7byY7vEB+6EvEyftGYm9Cib08MAAAAGAlIMkBAACAVS+1LyWv4KkwW1D+fF7RwagCP1B8OC4zZir0Q3m2JyfjFBMdWUeu5S4kO0IvVJhi5fGVyjDenIqs3EL2lc5bvL/SNQzDeMuxWusGAAAAViuSHAAArALNBugqHasUvKs1sAd0W3RNVNv+xTad+//OKT4SlxE1FLiBYkMxRWIRhWEo3ylOZeVkHNnztpyMIyfnqJApSNslRbp9F2iH0t+xMAyLa68sk4io5bxqf/+W/r0s7Vv6N5W/owAAAMDlSHIAALDCtSJAVy3QVinoRkAO/eLQI4f051v+XLlzOcmQ/IK/kOSQIQVeIM/2FqatKiU6PNuTbu1269FOpb9jpb+PzZ63VOlvaisSw1MnppQ+cWm9kX0pJSeXnyJu22frvrQk6fQ91cuevqf6dZZeo5Yyy5Wrp2yj16+njkbb1+n+aKYf6ynbbH9Wul61a9Tbp82+1516Fq1+R3vldw8AgGaQ5AAAYBVoNkDX6URFGFxKrpi1BwmBZg2ND+nm//Nmfft/+HZxeqq8V0xyxCPFILQfynM8eXlPTtZZWJ/j1//Dr+vvpv5OIp+HCiqNhmt0hN1ixw4d08njJxVbG5MMLbyPruVq1627dOjRQ403vk7bPlt/0LvW69ZbXyuuv/hYI8HndvRHtba2up2N1Nlsf9ZTfyPntVKr+7RcuWb6tBvvGgAAnUKSAwAANK2Raa6WOv2Pp/Vf/+y/Kv1sWpnpjCRpODmsif0TuuGPbtDbbnhbi1sNvNX+u/crEovoG/d8Q4X5ggbWDygyECkm3ALJd/3i2h3zBXkFTx/+iw9rzyf26O/u/7tuNx09bLm/i6VkcrUkcrWEyJmnz+jITUc0tGlIW2/cqvhwXIZZnG7NyTqyZ22devyUHhh4QIefOqzU/pSk6oHQWgKXlYKw1YKfpWONBK4XX7fW+mpVrV9qCT430r56+2O5ujrRzuXqXHrdaufWW1+jWtWn1TTap42Wa7ZPO/WuAQDQSWa3GwAAAPrLclNVLd6WJjXKHVvs2H9zTI/c8ojOPHNGg6OD2nL9FqUmU4oNxHT6ydN66CMP6bHbH2v7vQGSNPmpSX36+5/W2k1rlTub08VTFzX72qxmT89q7vSc7Iu2RpIjuvvFu7XnE3u63Vz0udL0gKW/j/VMc3Xm6TN66CMPKTmZ1PYPbNfE9RNK7UspsSeh8WvGtfGqjRrdPqqxnWMa2jykozcf1evPvN6uW5FUW4C8mYDo0rKtDK7WEhiu9xrt6I9Kiah2tbNa8qvetrTynpe7djeC7o2+m/WUa6ZPu/WuAQDQCSQ5AABAzdqx6O0Xdn9Br333NaWuS2nHr+7QtgPblNib0Pi7xrV5z2ZtfvdmjSRH9OO//bG+uPeLLa0bKGfTOzbp7h/drU889Qnd/L/frKt+7Spd9etX6YN/9kF94qlP6O6X79aGHRu63Ux0SK0JiHoTFUsTwaV9S69XzpGbjmjjjo1KTiaV2pfSlvdsUfK6pMbfOa4NOzZodNuo1k2s03BiWCPJEcXWxPTgjQ/W1LZeUmuAvRvTFC2ut9fat7S+Xm1nrXoxqN5on/b7s1iqX9oJAFi5mK4KAIBVoNyitvWc144Ex5f2f0n5C3lt/5XtGts5puHEsKKD0eJ0QHMFWectxYfixTURIoYu/vSivvLLX5FubmkzgLISexJK7Eno+n99fbebgi4p/U0s/V0sN31UtfOWXrOeupcrd+zQMQ2ND2ls55g2Xb1JicmERreNynd85c7lFB2MyjAMBV4g3/Hl2Z5cy5U9b+ux2x7TLX91S509gdWqkfUz2lF/t+rrxeQKAAC4HEkOAABWuFYG6JYLtlUK3lU6duKLJ3ThlQu68leu1PiucW2+drPWbVknSbLfsJU7l1MkHpGM4kLkgVsM1J39wVlps6R3N9QdAFC3ckmJpftrPa+eesqVPXn8pLbeuFVDm4c0MjGi0W2jWr91vexZW77jy7VcORlH8eF4cRuKKzYU0+CGQb346IttS3Ks1G90r9T7akQrkgD1rsXSzURDL7ShFryjAIDVjCQHAACrQDsDdA0dC6V/+tN/0sZf2Kj1b1uvDds3aPyd4xrbOSbrvKVMPKPADxa+eezmXDlZR07G0eDIoNx/dElyAFi1pk5MKbY2pvhwXLG1MZkxU77jy1AEP4QAACAASURBVJ615dmewuBS0jpiyIyaMmOmzLipaDxa3Aajmn5hWom9iabaUSmo2usB4Ub1wn2dvqfY98st+NzOQHe5azeTBGh0gft26WR/tkur16npxrsGAEC9SHIAAICOm/v5nDJTGW3YsUED6wc0sG5A0YGofMeXYRgyY6Yi8YgiA8UtOlAMykUHo4qujUo/lzTf7bsAsNK94x3v0NTU1MLPyWRSki7b18z+mZkZnTx5UjfddJMk6cknn6zp8/CPhyVDMkxDYRDKzbnKncvJd3yFQSgn68izPQVeoDAIpfDSyDpTMiLFMunn0k0nOVaLXv4mfzcCzc0Gu2vtz07fW7nFtUvtWC7Q3ys68Y6S1AAA9DKSHAAArGBr1qyR67oLP8diMUm6bN/i/ZlMRiMjI23/nH4uLTNSTGQYpiHf9ZW/mFcYhsURHJanwA3enI/eLAbzjOilbyWbpoJ00PoOA4BL7rvvPqXT6bqmkGp2NFxdn0MpcAO5WVf5i3lFB6NyLVeGYcizPRXmC3Jzrjzbk+/6xYSHHxaTHi1SLqBa7pvf/ahXExzlFqZu94LVlRIBtTzzehMcvdDvixMdvajdfdWtdw0AgHqY3W4AAABoj49+9KP6wAc+oJdffnlhK1m8b/H+L33pSx35PH9mXmbEVBiE8mxP9qyt7NmsMumMcudysmft4jeR8558x7/s28iSZBqmNNdQtwBAzVKp1GVbq/ePj4/rwIEDCz/X+jm1r3gNJ+soP5uXNWMpO5VV5vWMMlMZ5WZyyl/MqzBfkJNz5FqXJzsM01Bq/5vtaLWVEvzspUB7OafvuXzrZY30Zyl5snhbemw16+Q72k/vGgBg9WEkBwAAK9TevXvLHrv22muX3f+Zz3ymI583Xb1JvldcGLcwV5A1Y8mMmHKyzsLIDjdbXDTXtVz5BV++4xcDdH4gP/Cl8Yq3DwArVnIyKddyZc/ass5big5EFXiB4sNxmdE3E8hOzlFhriAneynRYXny7OKW2MNUVZX0Q4KjnF5sey+2qd/1Qp/2QhsAAJAYyQEAALogdV1KvuerMF9Q/kJeuXM5ZdIZZdIZZaeyyp3NybpgyZ61i99EvhSg821/Yc75MNW6KVcAoN/sunWXMulMcRTHdHbhb2hmKqPsdFa5mZys85byb+RVmCssjOqw52ztvm13t5tfl2ojQ1odaK33ep1uX6MabWez7W7k/peOGlhuBEGnRhRUGi3SbJ82+s70yjsFAECvIMkBAAA6bs3YGiV2J2Sds5SbyRUDdFOZN6daOZtT7txbEx2O5cjNuVJC0mC37wIAuufQo4fkWu5CUiOTvvQ3NH0pyXE2J2vGUv5Cvjh11VxB9pwtBdItD93S1ra1cwqhpddudV3NBo/b3b5q1210Ue9a27nceZXq7JdgfLX+lDrXp9XKdbpPez15BwCAxHRVAACgS37tc7+mv/zAXyp+Li4ZklfwFB+Ky4yZMozilFWe7cnNuipkLn0LOePIsz3pULdbDwDdd/ipwzp681FlzmTkWq5iQzFF41HJlEI/LP4dtbyFERxhGOqup+9qWf3VgrGVgt7V9i/3zffSOctdoxWB1qXrPZRTafHtcmWrfSO/2v56+rKSRttZrdxymunPZjTap40mpNrVp9Weeb192ul3DQCATmIkBwAA6Iotv7RF77z1nZr7+VxxwdxF01UtHc2Rv5CX/YYt+w1bu+/YLU10u/UA0H2p/Snd8c07lD2f1exrs5f9HS2N5sjN5JSZysjNurrz8TuVmGz/WhztmkaoUrC4F3SyfZXqqlZfo+1sps5eV6n93erTXrGSnzsAYOVgJAcAAOia3/xPv6nY2ph++Jc/lJt3FV8bV2QgIsM0FAbhm6M5cq6crKPJ357Uh/79h/T8/c93u+kA0BMmrp/QvYV7dfzjx/XSIy8pOhhVGBTXLDJMQ57tafdtu2ueoqqWoGUzgc1mg6LtDKq24tr1XqNbfdlo2XrX1WiHRhMHzV63XeU71afd/L0FAKDdSHIAAICu+o2/+A0l9yb1rT/4lvLn8zIihiLxiBRKvuMrCAIZpqFf//yva89/u6fbzQWAnnTw4YM6+PBBTb8wrfRzaUnFkR6JPe0fuQEAAAB0E0kOAADQdZOfnNS1/+paff/I93XqW6c0/cNpSVLi3QntuHmH9t61V2aMWTYBoJrE3oQSe0lsAAAAYPUgyQEAAHpCdDCqfb+zT/t+Z1+3mwIAWAHqWSyZ6XgAAAD6F1+JBAAAAAAAAAAAfYmRHAAAAACAFYfRGQAAAKsDIzkAAAAAAAAAAEBfIskBAAAAAAAAAAD6EkkOAAAAAAAAAADQl0hyAAAAAAAAAACAvkSSAwAAAAAAAAAA9CWSHAAAAAAAAAAAoC+R5AAAAAAAAAAAAH2JJAcAAAAAAAAAAOhLJDkAAAAAAAAAAEBfIskBAAAAAAAAAAD6EkkOAAAAAAAAAADQl0hyAAAAAAAAAACAvkSSAwAAAAAAAAAA9CWSHAAAAAAAAAAAoC+R5AAAAAAAAAAAAH2JJAcAAAAAAAAAAOhLJDkAAAAAAAAAAEBfIskBAAAAAAAAAAD6UrTbDQAAAACAXmYYxsLnMAwbOq8dxwAAAAAwkgMAAAAAyiolGUoJhsVJh1rPMwxDYRgubK04BgAAAKCIkRwAAAAAUEEpcVEt0VDuvEojMBo9Vs7UiSmlT6QlSal9KSUnk8uet+2zdV9aknT6nuplT99T/lgjZWtta6V6a1WurlqvvVz5WsouLdfp+tpVZ7P9Wel61a5Rb582816XK9+OZ9Hqd7Td/VLpOq34nQUAQCLJAQAAAAB979ihYzp5/KRia2OSIelSfsS1XO26dZcOPXqoY23Z9tn+DF5WCuaWjjUSfK7UH40mm6q1tdXtbKTOZvuznvobOa+VWt2n5co106fd6Jdq9fbr3woAQO8hyQEAAAAAHVKagqpVx848fUZHbjqioU1D2nrjVsWH4zJMQ4EbyMk6smdtnXr8lB4YeECHnzqs1P6UpOqB0FoCj5WCsNWCl/UENmsJErcqUFqtX2oJPi8+Xm9/1BuILpdYaFc7l6tz6XWrnVtvfY1qVZ9W02ifNlqu2T7tVL/UUr5biRcAwMrDmhwAAAAA0AHtSHA89JGHlJxMavsHtmvi+gml9qWU2JPQ+DXj2njVRo1uH9XYzjENbR7S0ZuP6vVnXm/pPS3Vz9/KbkVSZul5tSQMGg2q1xMAr3ZerVMWlTuv3ra08p6Xu3Y33sN6+7SRcs30aTf6pZH3BgCARpDkAAAAAIAKSutrVFv4u9J5rU5wSNKRm45o446NSk4mldqX0pb3bFHyuqTG3zmuDTs2aHTbqNZNrNNwYlgjyRHF1sT04I0PVryHftIr3wKvNZDb6fYura9X21mrXgyIN9qn/f4sAADoNUxXBQAAAABllBYRLyUuFiccFicgqp23+N/Fxxs9duzQMQ2ND2ls55g2Xb1JicmERreNynd85c7lFB2MyjAMBV4g3/Hl2Z5cy5U9b+ux2x7TLX91Syu6p2GNLrS9nF4Mfq8krZ4SrNH6u1Uf71dRowuW038AgE4gyQEAAAAAFZQbSbF0f63nteLYyeMntfXGrRraPKSRiRGNbhvV+q3rZc/a8h1fruXKyTiKD8eL21BcsaGYBjcM6sVHX2xbkqPZhbR7NSDKN+rf1IokQL2LiHfzveiFNtSiW8mgWvuF5BEAoJ1IcgAAAABAH5k6MaXY2pjiw3HF1sZkxkz5ji971pZnewqDS6NKIobMqCkzZsqMm4rGo8VtMKrpF6aV2Jtoqh2Vgqr1rB3QSHC2W4HnXgjMnr6neP/LLTDdzkB3uWs38ywaXeC+XTrZn+3Sjv5qpl/a8d4AALAUa3IAAAAAwDKmpqYu28rtf+aZZxaOPfnkk23/nD6RlgzJMA2FQSg35yp3LqdMOiPrvCUn68izPQVeoDAIpfDSlFemZESKZdLPpZvpmoZVW6C6F4PKvRyMLSU7SlsnlBawbnQh61r7s9PvQj++myXtfEdb1S/NvjcAAFRCkgMAAAAAlrj//vv1xBNPKAzDha1k8b7ljnXkcygFbiA36yp/Ma/sdFaZdEa5sznlL+RVmC/IzbnybE++6xcTHn5YTHq0yNKg5eLgZb0B4V4NevZqgqNcoLjdAeRKAe9annm9CY5e6PdeaEMl/TCqqdn3BgCAapiuCgAAAACW8corr+j2229/y/5UKlX25wMHDrT989S64qgSJ+soP5vXwMyADMOQk3FkRIqLjbs5V4X5gpycI9e6PNlhmIZS+y+/h1aqNJ1SK3QqqNtLgfZyerltSzXSn5UC4P3wfNqNPgAAoIgkBwAAAAD0keRkUq7lyp61ZZ23FB2IKvACxYfjMqOmwiCUZ3tyco4KcwU52UuJDsuTZxe3xJ7m1uNY6fo5eNyLbe/FNvU7+hQAgDcxXRUAAAAA9Jldt+4qrsExYy1MVZVJZ5SZyig7nVVuJifrvKX8G3kV5goLozrsOVu7b9vd7ea/Ra1T1nRiapt6g8fVpt3plWB0o+1stt2N3H+5qdAWX6NTaztUeuea7dNG35leeKeq/S52+30HAKwuJDkAAAAAoM8cevSQXMtdSGpk0hllXi8mOrLTWeXO5mTNWMpfyCt/sZjosOdsKZBueeiWtratWuC20v56Ewut1mzweOk9tisp02xCpdF2LndepTp7IRhfi1a8m63q02rlOtmnrfqdrfe9AQCgXkxXBQAAALSQYRgLnxcvGF3Pee04Vk/b0B8OP3VYR28+qsyZjFzLVWwopmg8KplS6IfyXV+e5S2M4AjDUHc9fVfL6m/km9y9vMjw4rbV8u39pftKZZYrW+0b+dX2t6ovG21ntXLLaaY/m9Fonzb6brarT6s983r7tNP9UrpWve8NAACNYCQHAAAA0CKlJEIpgbA4qVDredWOhWG4sNV6bLnj6H+p/Snd8c07lD2f1exrswtTVmWnsgujOXIzOWWmMnKzru58/E4lJtu/Fke5aYQqBbNrmXqo17/5XSmY3cm6qtXXaDubqbPXNftutqNPe0Gz/VLpGivhvQEA9A5GcgAAAAAtVEogLJdoqPW8Wq9Rq1KCAyvPxPUTurdwr45//LheeuQlRQejCoNLCTLTkGd72n3b7pqnqGomaNmq67ejbKeuX+81+q0v611Xox0aTRw0e912le9Un3arX1p1DQAAKiHJAQAAAPSJpUmPxYmLSsek2qeq+tk//Uzf+/Pv6cz3zmj+zLwkad3b1mnLL27RL/3BL2nLe7Y0fR9ovYMPH9TBhw9q+oVppZ9LSyqO9Ejsaf/IDQAAAKCbSHIAAAAAfWLpiIzFP1c6JqnisZLjHz+uf378n2VEDa3ZuEYTvzihwAtkX7B16tun9Oo/vKqrfuMqfeyrH2vH7aEFEnsTSuwlsQEAAIDVgyQHAAAAAP3Ha/+jstNZJa9LamjzkOJDcUmSm3dVmC8ofyEv67ylH/0/P9IXJ7+oTz3/qS63GKisnoWOmU4HAACgf5HkAAAAAFqoNEqi2loalc6r9Rqt8uXrvyzrvKVtB7ZpbOeYRpIjigxG5Bd8FeYKss5big/FFRmIyIgYuviTi3rwhgelD3akeQAAAABQFkkOAAAAoEVKiYlScqLcFFGVzqvl2OKfmz32/Jee14UfX9C2A9t0xbuu0Obdm7XubeskSfYbtnLncorEI5IhhUGowA3kF3xN/2BaukLSuxvoKKADGJ0BAACwOpDkAAAAAFqo3KLeS/dXWvy7Y8dC6ak/fUobfmGD1r1tnTZs36Dxd45r7B1jss5bysQzCvxAnu3JtVy5OVdO1pGTcTQwPCD3H12SHAAAAAC6yux2AwAAAAB0x9zP55RJZ7Rm0xoNjg5qYN2AooNR+Y4vwzBkxkxF4hFFBopbdCCq6GBxi62NSfOSkenMlFoAAAAAsBySHAAAAEALDAwMyHXdhc2yrIVjuVyuJz9PnZiSETEUiUdkmIZ811f+Yl6ZdEb52by8vKfADd6c7sqUDNOQETVkRk2Zpqnw9fKjRwAAAACg3UhyAAAAAE368Ic/rA9+8IN6+eWXF7Yvf/nLC8e/8IUv9OTnuZ/NyYyYCoNQnu3JnrWVPZtVJp1R7lxO9hu2nJwjL+/Jd3wFXqAwCKVLeQ3TMGXMMZIDAAAAQPewJgcAAADQpOuuu+4t+6699tqFz3/4h3/Yk583Xb1JgRfItVwV5gqyzlsyI6acrCPDNBS4wcIaHK7lyi/48h1fvusr8AMFQaBwEyM5AAAAAHQPSQ4AAABglUpel1TgBSrMF5S/kFdsbUyhHyo+El8Y4eHmXTkZR4X5gpzs5cmOIAhkTDCSAwAAAED3kOQAAAAAVqm1m9Zq87s2K3c2p9y6nMyYKd/1FZ+Py4yZUij5ji8n51yW6HByxU2bpXCQkRwAAAAAuockBwAAALCKfeizH9JXb/6qYmdjMmTIK3iKD8UViUckSYEbyLVduVlXhUyhmOjIOPIKnnSwy40HAAAAsOqR5ACADjOMN6f1CMPy334td16l8ouPVbs+AACS9Lb3vk3X3HKNfvSff6QwDOXa7ptJDkMK/VB+wS9OW5Utjuaw37D1rtvepe9v+f7CIuQAAAAA0A1mtxsAAKtJKQlRSj4sTUrUcl4YhhWTF6XjJDgAALX62Fc/pl237VLmTEaZ9Jtbdiqr7HRWuXM55WZyss5byl/Ma8+de/QvH/yX3W42AAAAADCSAwA6rZR8CMOwbJKjnvMaFbiBXvy/X9RPv/lTTf1gSpKU3JPUjg/t0O47dsuMkgcHgNXko1/+qCb2T+jv/+jvlT+flxExFqas8gu+Qj+UItKv/ftf09679na5tQAAAABQRJIDAFaYWqbD+uHRH+rrd39doUJFB6OKD8UlXzr1rVP68d/+WN/419/QR/6vj2jXbbs61WwAQA+47tPX6d13vlsvPPiCfvqNn+rsi2clSZtv2KwdH9qh6z55XXFBcgAAAADoESQ5AGAFWW6NjqX7vvWZb+mFL7+g8V3jGk4MKz4clwzJsz05GUf5N/KyZiz9zW//jaa/P633/+n7O3kLAIAuiw5Gtf9392v/7+7vdlMAAAAAoCqSHACwivyXT/4X/eivf6StN27Vxh0bNZwcVmxNTL7rqzBfUP5iXgMzA4oORhWJR/TM556RPW9Lm7vdcgAAAAAAAOCtSHIAQIeVRldUW2ej1vNqlX42rZcefklbb9iqK3ZfoSvedYXWTayTYRoqzBdknbcUWxuTGTGLC5f7oXzX1/f/0/elO0WiAwAAAAAAAD2HJAcAdFApaVFKXCyeSmrx1FLVzlv6uXR8aUJkcbmv/e7XNLp1VKNXjmrsqjFt3r1ZG3dslD1nKzudlQzJd315tic378q1XDk5R86cI+9vPem3W9kTAAAAAAAAQPNYNRAAOiwMw4Vt6f5az1u6lTtWkr+Y19kfntVwclhrxtYUt41rFBuKKbYmpuiaqGJrYstvwzFpSjIKrRlRAgAAAAAAALQKIzkAYBWYOjElM2YquiaqSCyiMAhlz9kyooYCN5Dv+AqDUDIkwzRkRAyZUbO4xcziea8H3b4NAAAAAAAA4DKM5ACADrj22mv1oQ99SC+99NLCVvKTn/xEn/vc5xZ+bsfn4188LjNiSqHkO8VFxnPncsq8nlHuXE6FuYJcy5Vf8BV4gcIgvGwKLNMwpZlmewEAAAC1Kk1dWss6buXOa8cxAACAXsNIDgDogI997GMqFArLHtu2bZvuuOOOhZ/b8fnAhw/o8a8/Ls/2VJgvKH8hr0gsIifryDAN+Y4vJ+PIyTpy85eSHW6gwA8U+mFxlMe6Bm8eAAAAdVm87lop2bB0CtNq5y0t04pjAAAAvYgkBwB0yMDAwLL74/G4Nm7cuPBzOz5fddNV+vvg74sJjot5xYfjCsNQ8UxcZtRU4AXy8sUEiJNx5OTeTHb4jq8gDGRM8E0+AACATiklFkoJjHrPq5SYaPRYOVMnppQ+kZYkpfallJxMLnvets/WfWlJ0ul7qpc9fU9911x8vXJlW11nLW1p5PrLla+l7NJyna6vXXU225+VrlftGvX2abPvWKeeRavf0UaeRT3PoZVlAfQHkhwAsAqs37pew5uHlb+Yl3XeUiQeke/6ig8XkxwKJa/gyc25xUTHvCM368q1XDl5RxoRIzkAAABWkEoJkVqTJccOHdPJ4ycVWxuTDEmXTnUtV7tu3aVDjx5qaZsr2fbZ5gLm3VKpLaVjjQSfK/VHo/dfra2tbmcjdTbbn/XU38h5rdTqPi1Xrpk+bVW/NHOdXvp9B9A+JDkAYBUwTEO//Ee/rMf/7eOKD8dlmIa8gqf4UFyReESS5Lu+vLwnJ+uoMF9YGNXhWZ70fikU0xQAAAD0o+WmnKo0JVW16arOPH1GR246oqFNQ9p649aF/74M3EBO1pE9a+vU46f0wMADOvzUYaX2pyRVD4TWEnyuFIStJ9FRj3Z/87tav9QSfF58vNb+KB2rNwhcLrHQrnYuV+fS61Y7t976GtWqPq2m0T5ttFyzfdpovwBArVh4HABWif1379fYVWOa/dmsMlMZZdKLtqmMcmdzys3kZF2wlL+Ylz1rK38+r/Fd49J13W49AAAAGtHqNTXOPH1GD33kISUnk9r+ge2auH5CqX0pJfYkNH7NuDZetVGj20c1tnNMQ5uHdPTmo3r9mddbVv9yGvlmfq9MWVNLYLjea9SSMGg0qF5PALzaebVOxVTuvHrb0sp7Xu7a3Xin6u3TRso106et6Jdmfmd77fcdQPuQ5ACAVeRTz31KI4kRzb46e1mSIzuVVXY6q9zZnKwZS9YFS9lzWa1/+3p98nuf7HazAQAAVp3SlFGV1uOodl61BcsrXa+cIzcd0cYdG5WcTCq1L6Ut79mi5HVJjb9zXBt2bNDotlGtm1in4cSwRpIjiq2J6cEbH6x4zU5bCQHPWhMAnf7m/NL6erWdterFd6XRPu3nZ9HMc+jFZwig9ZiuCgBWEcM0dPfLd+uRjz2i1777mqyzlqJro4oMRIrrctie3LwrM2Zq52/s1K3Hbu12kwEAAFad0iLipYRDuemjqp23+N/FxystUl7p2LFDxzQ0PqSxnWPadPUmJSYTGt02Kt/xlTuXU3QwKsMwFHiBfMcv/rel5cqet/XYbY/plr+6pfnOaUKr1qIgaNp+3f4GfreTQ7xjRazFAaBWJDkAYBX6rf/8W3rtO6/pn/6Pf9LUc1PKpDOSpLVja7XtX2zTDf/mBl154MruNhIAAGAVKzfFVKW1NWrZX8vxcsdOHj+prTdu1dDmIY1MjGh026jWb10ve9aW7/hyLVdOxlF8OF7chuKKDcU0uGFQLz76YtuSHLUEM1sZNO9UAJ4g7ZtakQSodxHxbiYaeqENtWjnO8o0VQDqQZIDAFapK993pa5835WSJC/vSZKia/ifBQAAALzV1IkpxdbGFB+OK7Y2JjNmynd82bO2PNtTGFwaVRIxZEZNmTFTZtxUNB4tboNRTb8wrcTeRFPtqBRUbWTx6mrKLbLdSb0QqD19T/Hel1tguhOB7nL7W/FMGz2nVXrhHWtWuxdy73RZAP2HNTkAAIquiZLgAAAA6AFTU1OXbZX2P/nkkwvH2/35uw9/VzKK05+GQSg35yp3LqdMOiPrvCUn68izPQVeoDAIpfDSVFmmZESKZdLPpRvtlqY0GjSutrB1r36Lvd1KyY7S1gmlBawbXci61v7sdIKhm+9Ys9r5jjJNFYB6keQAAAAAAKAH3H///XriiScUhuHCVrJ43+K1NRYfb+fn4g4pcAO5WVf5i3llp7PKpDPKnc0pfyGvwnxBbs6VZ3vyXb+Y8PDDYtKjRZYGuxcHvZcLbrYjENupaap6LcFRLsHQaOKhnnrL7WvlVGW91O+90IZKOpHgYJoqAPXga7sAAAAAAPSIV155Rbfffvtb9qdSqbfsO3DgQOc+335Ar37hVTlZR/nZvAZmBmQYhpyMIyNSXGzczbkqzBfk5By51uXJDsM0lNr/1ntolUrTKUmVg+G9FBjtpbaU08ttW6qR/uyXd6VbemFNmmpt4BkCqw9JDgAAAAAAUFFyMinXcmXP2rLOW4oORBV4geLDcZlRU2EQyrM9OTlHhbmCnOylRIflybOLW2JPc+txrHT9HHztxbb3Ypv6HX0KoFeR5AAAAAAAAFXtunWXTj1+qpjYiBQXHo8Px2XGTCmUfNeXa7lyssVER2lUhz1na/dtu7vS5krB2EYDtu2Y87/ettQ6cqXbwehG21kq16hG7r8d70qjKt17s33a6DvTqT5o5jn00jME0FmsyQEAAAAAAKo69OghuZarzFRmYT2OzOsZZdLFn3Nnc7JmLOUv5JW/mFdhriB7zpYC6ZaHbmlr21qdeCh3vcX7WxUsbTb4urSt7Vp4uVqf1Luod63trHetlX4JZrfiHWtVn1Yr1y99CmD1YiQHAAAAAACoyeGnDuvozUeVOZORa7mKDcUUjUclUwr9UL7ry7O8hREcYRjqrqfvaln91YKxrQzCtitZUK6Oat/eX25fqcxyZetdr2Dp/nrXNimn0XZWK7ecZvqzGY32aaPvWLv6tNozr7dPm3nXAKAejOQAAAAAAAA1Se1P6Y5v3qHs+axmX5stjuZIZ5Sdyi6M5sjN5JSZysjNurrz8TuVmGz/Whyn72ltoLTStVpdVzMambanHXVVq6/RdjZTZ69r9h1rR58CQL9iJAcAAAAAAKjZxPUTurdwr45//LheeuQlRQejCoNQkmSYhjzb0+7bdtc8RVUtwdVOBu3bWWc76qj3Gs3U2Y2yrVpXoxnteleabW+v92mrn0cvtQVAbyHJAQAAAAAA6nbw4YM6+PBBTb8wrfRzaUnFkR6JPe0fuQEAAFBCkgMAAAAAADQssTehxF4SGwAAoDtIrSynNQAAIABJREFUcgAAAAAAALRAPQtJM30OAACtwcLjAAAAAAAAAACgLzGSAwAAAAAAoAUYnQEAQOcxkgMAAAAAAAAAAPQlkhwAAAAAAAAAAKAvkeQAAAAAAAAAAAB9iSQHAAAAAAAAAADoSyQ5AAAAAAAAAABAXyLJAQAAAAAAAAAA+hJJDgAAAAAAAAAA0Jei3W4AAAAAAACNMAxj4XMYhnWfV618peO11g0AAID2YiQHAAAAAKDvlJIMpQTD4qRDreeFYVg2QWEYxsLxMAwvK1fpGAAAADqLJAcAAAAAoC+VEhTVRlLUet5yZQAAANDbmK4KAAAAAIA2eePUG3r288/q1Sde1cVTFyVJG3ds1PZf2a7rf+96jV452uUWAgAA9DeSHAAAAAAAVFCanqqk1imqHv83j+v5Lz8vhdLg6KA2XLlBgR/IOmvpxBdP6IUHX9C+39mn9/+797ez+QAAACsaSQ4AAAAAAMpYmuAoKbeAeclXf/WrmjoxpcSehIY3Dys+FJckubarwnxB9kVb1nlL3/vc93T2hbO6/Ru3t+8mAAAAVjCSHAAAAACAvlRKQFQbVVHreeXK1XvO0ZuPavoH09p2YJvGrhrTcHJY0cGofMdXYa4g64Ila8RSZDAiI2ro9FOn9dCHH5L219U8AAAAiCQHAAAAAKAPlZIWpcTF0pEVixcbr3Te0s+l46WfF5+z9NjS60nSy8df1pn/94y23rhVV7zrCm3evVnrtqyTDMmetZU7l1N0MCrDNBQGoQI3kO/4eu27r0nrJO1sqlsAAABWHZIcAAAAAIC+VG6UxdL9tZ7XimNP/M9PaMOVG7R+63pt/IWNGn/XuMZ2jil/Ia9MOqMwCOU7vty8K9dy5eQcOVlHhaGC3H9wSXIAAADUyex2AwAAAAAAWAmyU1ldPHVRa69YqzUb1mhgdGBhLQ4zaioyEFF0MPrWbU1UsaGYdEEycvVNqQUAALDaMZIDAAAAAIAWSJ9ILyQzjIihwAtkz9qSIQVeIM/2FHiBFBanvDJMQ2bElBktbkbEUJiuvAYIAAAALsdIDgAAAABA33jyySd14sSJy7aS+++/v6ufZ/95VmbELE5JVSguMp6dziqTzsg6Z6kwW5Cbc+XZnnzXV+iHCoNwIekRMSLSxfr7pN+U1kipZcH4cueVO7Z4f70LzQMAgP7ESA4AAAAAQN/47ne/qzAM9Sd/8idvOXbfffd19fPo20eLIzbyngrzBVnnLZkRU07WkRkx5Tu+nGxxDQ7XejPZEXiBAi+QH/jSxpq7oi8tXuC9lIhYbo2TSuctLbP050prpgAAgJWHJAcAAAAAAC2Qui6lwA9UmC8ofzGv2NqYwiBUPBOXGTUV+qHcvCsn4xS3nCPP8orJDsdXGIQKUys/QF9KQpQSGPWe1+okxtSJKaVPpCVJqX0pJSeTy5637bONXf/0PdXLnr6ntmstd53lytba1lrrraRcXa2+p2rlOl1fu+pstj8rXa/aNert02bf6049i1a/o/U8i069N8BqR5IDAAAAAIAWGE4Oa2zHmPLn88qtz8mMmvJdX/HhuCKxSHEaK8eXk3PkzF/aLo3qcPOuNCZpqNt30R8qJT1qTYgcO3RMJ4+fVGxtTDIkXTrVtVztunWXDj16qKVtrmTbZxsL0tZStp2qtUtq7L4q3VOjyaZm+rCRdjZSZ7P9WU/9jZzXSq3u03LlmunTZvqlHe9bLWWB1YokBwAAAAAALfK+f/c+Hb/9uGIjMRmGIa/gLSQ5JClwA7m2KyfrqDBfkD1vy8k68vKe9LEuN76PlJuuarmEx9J9Z54+oyM3HdHQpiFtvXGr4sNxGaahwA3kZB3Zs7ZOPX5KDww8oMNPHVZqf0pS9UBoLYHHSkHYWoKXtQZxawmgtipQWq1fagk+Lz5ea3+UjtUbiC6XWGhXO5erc+l1q51bb32NalWfVtNonzZartk+bbRfytXdzEirbiSkgH7AwuMAAAAAALTIOz76Dl1505WaOz2nzFRGmXRGmdcv/TuVUfZsVtaMpfyFvOw3bBVmC7IuWHr7B94uXdXt1q98Z54+o4c+8pCSk0lt/8B2TVw/odS+lBJ7Ehq/Zlwbr9qo0e2jGts5pqHNQzp681G9/szrbW1TrVP6lDuvm9/qriUwXO81akkYNBpUrycAXu28djy3VvRnrfUvvXY33qN6+7SRcs30aaP90szvbC//vgO9jCQHAAAAAAAtdPvXbtfW927V3M/mlE1niwmOdEbZqayyZ7PKnc0pN5OTdd5S7nxOv/D+X9Btf3Nbt5vdMaXppCqtx1HpvGrlKjly0xFt3LFRycmkUvtS2vKeLUpel9T4O8e1YccGjW4b1bqJdRpODGskOaLYmpgevPHBhuvrNb3yLfBaA7mdbu/S+nq1nbXqxYB4o33a788CQHsxXRUAAAAAAC12xzfu0D/88T/oub94TtmzWUUGIooOFP8vuGd78gu+ImsiuuGPbtCB+w90t7EdVFpEvJSoqDTtVLnzKi1EvjQBsvjYsUPHNDQ+pLGdY9p09SYlJhMa3TYq3/GVO5dTdDAqwzAUeIF8x5dne3ItV/a8rcdue0y3/NUtLeyJ2rR6aqmSXgx+ryTtem711t+t+lbz+9XMs+/2ewP0M5IcAAAAAAC0wfv/1/dr36f26Zm/eEav/cNruvCTC5KkjTs36u3vf7uu/++u17ot67rcys4rtxj40v2VFg2v9RqLnTx+Ultv3KqhzUMamRjR6LZRrd+6XvasLd/x5VqunIyj+HC8uA3FFRuKaXDDoF589MW2JTnqCUj3UzCZb9S/qRXPrd5FxLv5bvRCG2rRiXe0mWffT7/vQLeR5AAAAAAAoE3Wb1uvX/3ffrXbzVj1pk5MKbY2pvhwXLG1MZkxU77jy5615dmewuDSyJGIITNqyoyZMuOmovFocRuMavqFaSX2JppqR6Wgai3z9JfbX0vws1uB514IzJ6+p3j/yy0w3c5Adyue21KNLnDfLp3sz3ZpR3818+zb8d4AKx1JDgAAAAAA0HZTU1OX/ZxMJuva/8orr+imm26SJD355JN1ff7uw9+VDMkwDYVBKDfnKncuJ9/xFQahnKwjz/YUeIHCIJTCS1NfmZIRKZZJP5duOsnRrH4JKPdyMLYbfdbsc6u1Pzt9b+UWTy+1Y7mkUq/o1DvazLPvl993oBew8DgAAAAAAGir+++/X0888YTCMFzYShbvK7e/9PPiY/V8Lu6QAjeQm3WVv5hXdrq4KHzubE75C3kV5gtyc25xzRTXLyY8/LCY9GiR0/csv0nVA5jlAsq1lO2kXk1wLO7rWva3st5y+2p5bvUmOHqh33uhDZV0K8GxeN9K+X0HegUjOQAAAAA0rdwiwLWeV6l8tWvXWjeA7nrllVd0++23v2V/KpVa9vyl+0sjPCTpwIED9X2+/YBe/cKrcrKO8rN5DcwMyDAMORlHRqS42Libc1WYL8jJOXKty5MdhmkotX/5drZCpemUWqFTQd1eCrSX08ttW6qR/qwUAO+H59Nu9AGwMpHkAAAAANCUUpIhDC/NaW8YFZMRy51X+ndxwqKk0rGldZWrG8DqlpxMyrVc2bO2rPOWogNRBV6g+HBcZtRUGITybE9OzlFhriAneynRYXny7OKW2NPdqap6XT8Hj3ux7b3Ypn5HnwIrF9NVAQAAAGjactPJNHNevfXW6uwPzurZ//Cs/vq2v9Zf3/bXevbzz+rsD862pC0AetuuW3cpk87ImrEWpqrKpDPKTGWUnc4qN5OTdd5S/o28CnOFhVEd9pyt3bft7kqbWxGM7cTUNvUGj6tNu9MrwehG29lsuxu5/3JToS2+Rrun5iqp9M4126eNvjPdnKKqE2WB1Y4kBwAAAIC+VhoVUmkUx/kfn9fnr/m8vvLer+jb//bbevU7r+rVb7+qx//ocX3lvV/R56/5vN746RsdbjmATjr06CG5lruQ1MikM8q8Xkx0ZKezyp3NyZqxlL+QV/5iMdFhz9lSIN3y0C1tbVs9azMst6+W4Gi7AqjNBo+X3le7kjLNJlQabWe9z61XEjzVVOtPqXN9Wq1ct/q0md/ZZn/fgdWG6aoAAAAA9LVq01U9/8Xn9c3f/6bWb1uvrTdsVXw4LsMw5Du+CpmC7Flb2emsPr/78/rwX3xYez6xp9O3AKBDDj91WEdvPqrMmYxcy1VsKKZoPCqZUuiH8l1fnuUtjOAIw1B3PX1Xy+pvdLHhUrleW3B4cXtq+fb+0n2V7qvaN/Kr7a+UQKhHo+1s5Lk105/NaLRPG30f29Wn1Z55vX3aaL808zvby7/vQC8jyQEAAABgxXr288/qO//Td7Tll7Zo446NGk4MK7qmOBe/k3WUv5iXdd5SbG1M8bNxfeOeb8h3/G43G0CbpPandMc379CDNz4oe97W4IZBReNRGRFDYRAq8AJ5trcwguOup+9SYrL9a3FUC1ovDnzWU67Xv/nd6H11uq5Gy3by/jqt3L2VjjVafqX0aTfeN2A1I8kBAAAAoGmlERTLLQ7eyHn11ruc3ExO3/rMt/S2X36bNl+7WePvHNfIxIjMiCkn68i6YCk+FFckHile51KA8+u/93XpM5IGW9JEAD1m4voJ3Vu4V8c/flwvPfKSooNRhUHx74hhGvJsT7tv213zFFWdmiqqkWv08voDjV6jW2seNFq23nU12qGWxEE7rtuu8p3q027dXyvqBlYbkhwAAAAAmlJKWpQSF+Wmj6p23tLPpeOVji1NmCy+5vHfOq6RLSMa3T6qsXeMafOezdqwfYNcy1V2OiszZioMLk1PY3ty865cy1X8jbjsY7b0r1rQOQB61sGHD+rgwwc1/cK00s+lJRVHeiT2tH/kBgAAaB2SHAAAAACaVm40xdL9tZ5X67Fyx728p9f+8TVte+82DY0PaeiKIY0kRrR201rZs7acnKNYJqbYUEyxtZdvA+sHZL9qS8xaBawKib0JJfaS2AAAoF+R5AAAAACw4qRPpBUdjBYXFR6MyjAMOTlH1owlr+ApcAMpLI4MMSOmzKipSCxS3OIRGVFDSnf7LrCalRuhVM95jRxbOpVctSQjVpZ6FjpmOh0AQK8wu90AAAAAAP0pkUjove99r06dOrWwSdLXvva1hXMeeeSRrnz++oNfl2mYkiH5ri8n6yh3Lqf59Lys85YK8wV5eU++4yvwAymQFEoyinPym6YpTdfbI0BrVJqyrdbzSlPFlbZap4Qr7S9tAAAAvY6RHAAAAAAa8ulPf1qzs7Nv2X/NNdcsfH73u9/dlc9XXn2lng+el1/wFxYZj8QjcnKODMOQX/BVyBTk5lx5eU+e48n3fIV+qDC4FNxl4XF0Ubl1Z2o9r9Zp3qpdvxZTJ6aUPnFpTYt9KSUnk01dD93D6AwAQD8iyQEAAACgYaOjo2/Z9/a3v33h89VXX92Vz/s+sk8n/uSECpmC7Dds5c7lJEnxTFxm1FTgBXItV4X5gpysI9dy5dmevMKl0R1hIKVq6ABgBao1WXLs0DGdPH5SsbUxyVBxNJQk13K169ZdOvTooTa3FAAAgCQHAAAAgBVobOeYItGI7DdsWTPFURyBFyg+HFckFlEYhPJsT07OKSY6Mo7c7KVRHbYnmVK4kal6sDKUpq6qxXJrdyzdd+bpMzpy0xENbRrS1hu3Kj4cl2EaCtxATtaRPWvr1OOn9MDAAzr81GGl9pMxBAAA7UOSAwAAAMCKY0QMTX5yUie+dGIhAOsVvIUkh1Rcq8OzPBWyBRXmCipkismOQqYg7VPxm+lAn6snwVGLM0+f0UMfeUjJyaTGdo5paPOQYmtjCoNQbtZVfjYv67yl+HBcmamMjt58VHd8846W1Q8AALAUSQ4AAAAAK9IH/+yDevGhFzX/83mFYXHkRmw4VkxyGFLohfIKXnHaqkwx0ZF/I6+1G9dq/lfnF6beAbqhlJyotl5GpfMqJThqvf5SR246ouRkUsnJpDZdvUkjEyMyY6bcnKv8xbwGZgYUHYjKjJiSpMyZjB688UHpf6yrGgAAgJqZ3W4AAAAAALTL7770uzIihuZOzymTzijzekaZdEbZqayyZ7PKncvJmrGUv5BXbian2FBMv/PS73S72VjlSomJUgJicaJiufUyKp1nGMbCVmu5xdviY8cOHdPQ+JDGdo5p09WblJhMaOL6CV2x6wqNXjmqkdSIhq4Y0tpNa7Vm4xoNjg5qcOOgovGodKwFHQMAALAMRnIAAAAAWLHWjK3R3T+6W1/a/yVlXs8oMhBRbCgmI2JIoRR4xTUEAjfQuret0yef/aTiw/FuNxsoOwJj6f5az2v0+oudPH5SW2/cqqHNQxqZGNHotlGt37pe9qwt3/HlWq6cjKP4cLy4DcUVG4ppcMOgnJNOxfYAAAA0ipEcAAAAAFa0wfWD+r1Xfk8H/uSAYkMxzf1sTtaMJeu8pfkz8xpYP6D3/S/v090v302CAyhj6sSUYmtjig/HFVsbkxkz5Tu+7Flbnu0pDIpTXxkRQ2bUlBkzZcZNRePR4kiOqGRMs9ANAABoPZIcAAAAAFaF9/zBe/T7p39f9wX36c7v3Kk7v3On7gvv0++/9vv6xf/+F7vdPECS9I53vENTU1MLW8nifS+//PLC/ieffLIjn9Mn0pIhGaZRXGQ85yp3LqdMOiPrvCUn68izPQVeoDAIpfDSdFimiokPw5TSDXcLAABAWUxXBQAAAGB1MaTU/lS3WwG8xX333ad0Or3slFG1TC/V7s8KpcAN5GaLi4xHB6NyLVeGYcizPRXmC3Jzrjzbk+/6xYSHHxaTHgAAAG1CkgMAAAAAgB6RSi2fgFu8f/HnAwcOdORzal+xTifrKD+b18DMgAzDkJNxZEQMBV4gN+eqMF+Qk3PkWkuSHWGoMEWyAwAAtB5JDgAAAAAAUFFyMinXcmXP2rLOW4oORBV4geLDcZlRU2EQyrM9OTlHhbmCnOylRIflFdfs8EIp0e27AAAAKxFJDgAAAAAAUNWuW3fp1OOniomNSHHh8fhwXGbMlELJd325lisnW0x0lEZ12HO2tKvbrQcAACsVSQ4AAAAAAFDVoUcP6YGBB5SZykiSPNtTfCguM27KMIpTVnm2J9cqTltVmCsUExyBpEOSmK0KAAC0gdntBgAAAAAAgP5w+KnDcrOuMmcyyqQzmk/PK5vOKjOVUXY6q9xMTtaMpfzFvKyLlsIw1F1P39XtZgMAgBWMJAcAAAAAAKhJan9Kd3zzDmXPZzX72qwy6WKyIzuVLSY5zuaUm8kpM5WRm3V15+N3KjHJYhwAAKB9mK4KAAAAAADUbOL6Cd1buFfHP35cLz3ykqKDUYVBcS4qwzTk2Z5237Zbtzx0S5dbCgAAVgOSHAAAAAAAoG4HHz6ogw8f1PQL00o/l5ZUHOmR2MPIDQAA0DkkOQAAAAAAQMMSexNK7CWxAQAAuoMkBwAAAAAA6BjDMBY+h2HY0Hnlji3eX085AADQv1h4HAAAAAAAdEQpyVBKMCxNStRynmEYCsNwYVsusVHall6zUjkAANCfGMkBAAAAAAA6ppR8qJZoKHdeoyMwGik3dWJK6ROX1hvZl1JyMrnseds+21CTdPqe6mVP31P+WLmylcrUco16ytd77Xqu32jblpbrdH3tqrMVz7vc9apdo94+bea9Lle+Hc+i1e9oo/3S6L3Vew1gpSLJAQAAAAAA+kojU1lVO7bYsUPHdPL4ScXWxiRD0qVTXcvVrlt36dCjh5pofX22fXb54GWlgGfpWCPB3Up11qNd7avUtkaTTc30RSPtbKTOZvuznvobOa+VWt2n7fgdaqRfWv0MAbyJJAcAAAAAAOgrS9fhWDzqY7HFxyqVKznz9BkduemIhjYNaeuNWxUfjsswDQVuICfryJ61derxU3pg4AEdfuqwUvtTkqoHQmsJXFYKwpYL0lart5ZkRb3B33o02r5yIwxqva/SsXrvo1xioV3tXK7Opdetdm699TWqVX1aTaN92mi5Zvu03n5p1+8ssNqxJgcAAAAArGCGYSxsjZ7XjmNArznz9Bk99JGHlJxMavsHtmvi+gml9qWU2JPQ+DXj2njVRo1uH9XYzjENbR7S0ZuP6vVnXm9rm2oJrtZ7TKqefGlFELWZ9pU7r5bgb6NB9XoC4NXOa0f/t6I/a61/6bW7EVSvt08bKddMnzbSL618hgAuR5IDAAAAAFaodi/y3Mzi0Fi9Su9CLYm35c5r9F2qVu7ITUe0ccdGJSeTSu1Lact7tih5XVLj7xzXhh0bNLptVOsm1mk4MayR5Ihia2J68MYHG2oLyqs1AdDpaZSW1ter7axVLwbVG+3Tfn8WAJrHdFUAAAAAsIK1e5HnWq9fqwuvXJAkje0ca/pa6D2l92Rpgkx667RT5c6r9I4ufQdrLXfs0DENjQ9pbOeYNl29SYnJhEa3jcp3fOXO5RQdjMowDAVeIN/x5dmeXMuVPW/rsdse0y1/dUtT/dJJzP3fXd3u/24nh3jvmkefAm9FkgMA/n/27j9GjjO/8/unarp7huwhOeSQy54e7XC5lqj10oxJLqls1kuT2V2f7EM2hskRopWCk0HBQe5yB8FBnCC5KFoBOgMJEBi6+GwjsUUYx1BeEVxcEjirBc/eZaBwczrRvEQKHcumJdpU90j8oRl2d3V1VXVV/ujp0ajVP6qrf1XPvF9AQz1V9VQ9/a1qYvf77ed5AAAAMFLX/6fr+slv/UT3/uqeEqna/031HE+zD8/qa//Z13T47OER9xD91Kpw1ri9XYEt7DnC7r9x6YYWTiwovTetbfPbNLNvRjsWdshetlV1qnItV07BUWo6VXulU0qmk5raOaW3X3t7YEWOQSyk3enYYSRM+UX9J/oR/24XER9lUjwOfQhj1MWgKG3jHlNgkChyAAAAAABCabZQcythRnZ4FU+//2//vlb+dkVbdm7R/LF5mSlzbZHn0p2Sfvif/lBv/rM39Wtv/pqMCaa8Qv/lr+WV3JpUajql5NakzKSpqlOVvWzLsz0F/uqokglDZsKUmTRlpkwlUonaayqhpetLyhzJ9NSPdknOqMnLdu1aXW+YCdM4JGVvPVf7zM0WfB5konsQ8Y+6wP2gDDOegzLsZ7TbRcfHMabAILAmBwAAAACgo24KHHXr1+RoVClU9E/3/1NVliva9/V9+uI3v6j5r84rczijz/3M57T7S7v1uS9/Tju+sEMfv/exfmvht+SUnH59HIzAo48+qnw+v/aqW7+t3fY333xzbd+VK1f69j53LScZkmEaCvxAbslV6aOSCrmCrLuWnKIjz/bke74CP5CC1WmxTMmYqLXJvZXrQ4T6o9skeX0B5WEtMB3nX53Xix311zD0Gv+w8Rx2MrzT4ulxTs4P+xkNe71xjikwaIzkAAAAAIANrF6cCLPIc6vj2hU4wpy/Wfvf/ZnfVTKd1ENffUizj85qW3abJlIT8mxP9rIt665V+1V9ypRhGFr52xX93qHfk54J8aEROy+88IJyuVzT5yjK9FV9fx9IvuvLLboq3y8rMZWQa7kyDEOe7anyoCK35MqzPVXdaq3gUQ1qRY8+aZXgbDXKoNWx7c4V5prtRjb0Kq4FjlYLUw96wepe499tgSMOca9/vriKa4GjnbjHFBgGihwAAAAAsEH1Y5Hn+rZmCzaHade4XZL+9L/+U1UeVPTFb31Rew7uUeZnM9r++e0K/EDle2UVPyrKTJi1xHN9oeeKp5VbK9KPJZ3sNTIYhWw229P29X+fOnWqb+/z22ujR5yio/JyWZN3JmUYhpyCI2Oitti4W3JVeVCRU3LkWp8udhimoezx5p+hH8ImveOUyG4m7v2T4t23RlHi2S4RPg73Z9DGscABoIYiBwAAAABsYL0u8hx1IedW233P11u/95ZmD8yuLfA8++isdn5xp8r3yzJMo1bUsDy5pdpiz5VCRanplCZ3TMp905Xx86zNgf6ZOzon13LXRhAlJhPyPV+p6ZTMhKnAD+TZnpySo8pKRU5xtdBhefLs2itzuLf1OHoV92Rp3PvXThz7Hsc+jTsKHMB4Y00OAAAAAMDQfPzXH8uzPE3tnNLktkkltyZlGIZcy60t8GwatYWdk6YmUhOamJxQYjKhxGRCyS1JyZWCj/s3RRAgSQefOFhbg+OOpeJSUYVcofbKF1RcKqp0pyTrrqXyx2VVViprozrsFVuHnjw00r73Y4qqQYravzgsjN5O1H722u8on79x3Y9ma4AMe02WVv1sd0ynmEZ9Zsa9wMFUVQBFDgAAAADYkAqFwqdezbZXKhXdunVrbd8w3uev5SVDmkhOSJK8iifrnqVCrqDyvbLckquqU1Xgry5YvrogtDFhyEyYMg1Tis8az9ggFl9blGu5a0WNQq6gwge1QkdxqajShyVZdyyV75VVvl8rdNgrtuRLpy+cHmjfBjnFULNz9zMB2+/+DSqZ22tBJWo/u41/XAo8nXSKpzS8mHZqNy4Fjn7EFNjImK4KAAAAADaYP/zDP9T+/fuVyXwyhc62bdskSfl8fm3bzp079aMf/Ui/+qu/KklDef/lwpdlmKvrHJRd2R/bKi4V1xZ5dsu1Kaq8sqeqU5Xv+vKrvuTXpsAyZcq3/L7GC5Cks2+c1fnHz6twuyDXcpVMJ5VIJSRTCqqBqm5tGrX6CI4gCPTs1Wf7dv1OydjGBOb648P8Or5xW73NMAoH/e5fp1/kd9reroDQjaj9jBL/XuLZi6gxjfpcDSqmne55tzGNEpde7yEjNoDWKHIAAAAAwAbz/vvv67333tN3v/vdz+w7cODAp/6uFyCG9f7WlVsK/KC2vsFyRaU7JRmmIafoyJwwVXWqtX0PKnJLqws8O1VV3aoCL1A1qCrYy3RV6L/s8ayefv1pvXLiFdkPbE3tnFIilZAxYSjwA/meL8/21kZwPHv1WWWODn4tjkGmHLaNAAAgAElEQVT9Ont9UngY1+vWMPvXy7Wito17/HvR6rPV90VtT0xb7wM2O4ocAAAAAIChmfvKnKpOVZWViqx7lhJbEgr8QKnC6iLP1doiz5XC6roH9UWebU9exVNQDWTMs/A4BmP+sXk9X3lel75zSe987x0lpmrPp1SbNs2zPR168lDoKarCJnSj6Edic5DJ0VH0r5drjqJtt+tqDEKYwsEgzjuo9sOKaZS2o4oJsBlQ5AAAAAAADE1qOqXP/zuf172/vKfJbZMyE6aqblWpdEpm0pQCqepU5VpurdCxUpFTcOSUHDlFR1qQggQjOTBYZ149ozOvntHS9SXl3qotApM9nlXm8OBHbgAAgO5Q5AAAAAAADNWvnP8V/faB31bxo6Jk1BYfT6VTmkjVFiOvTwtUn7aqXujwbE86M+LOY1PJHMkoc4TCBgAAcUaRAwAAAAAwVDsWdujnX/h5vfGbbyjwA7ll95Mih7G6yLNTrS1CXlwtdBQq+sY/+YYuly5LDOQAhq6bRY+ZVgcAMEwUOQAAAAAAQ3fivzyh5FRSf/qP/1ROwdHktklNTE7IMA0FQSDfXR3NUXDk2q7+zn/3d3T8Hx7X5Rcvj7rrAAAAiBGKHAAAAACAkfjqr39VCz+3oIv/wUUVl4qqelVNJGtTVlWd2vv07rT+3r/8e8ocZcogYJQYnQEAiCuKHAAAAACAkck+ltVzf/2c/ub//Bu99yfv6W+v/q0k6fM/93nt/8Z+LXx9YcQ9BAAAQJxR5AAAAAAAjJYhLXx9gYIGAAAAumaOugMAAAAAAAAAAABRUOQAAAAAAAAAAABjiSIHAAAAAAAAAAAYSxQ5AAAAAAAAAADAWKLIAQAAAAAAAAAAxhJFDgAAAAAAAAAAMJYocgAAAAAAAAAAgLFEkQMAAAAAAAAAAIwlihwAAAAAAAAAAGAsUeQAAAAAAAAAAABjiSIHAAAAAAAAAAAYSxQ5AAAAAAAAAADAWKLIAQAAAAAAAAAAxhJFDgAAAAAAAAAAMJYocgAAAAAAAAAAgLFEkQMAAAAAAAAAAIylxKg7AAAAAAAAPs0wjLX3QRB0fVyn9lHbAQAAxA0jOQAAAAAAiJF6oaFeZFhfeAh7XBAELYsUhmGs7Q+CIHQ7AACAOGIkBwAAAAAAMVMvNDQWIaIe16xNv+Sv5ZW7lpMkZY9lNXd0rulx+16Odv5bz3Vue+u57s65/nzdtI3aLuw51wt7/mbtw7RtbDfs6w3qmr3Gs935Op2j25j2+lwP6170+xmNci/C3od+338A44EiBwAAAAAA6NrFxYu6cemGkluTkiFptXbiWq4OPnFQi68tDq0v+17uLWE+yHZRz1nfFyWh2y4eg/j8neIfpZ9RrtlrPLu5fpTj+qnfMW3VrpeY9isu/bgPUe4/gPFBkQMAAAAAgE2qPnVVN25fva1zJ88pvTuthRMLSk2nZJiGfNeXU3RkL9u6efmmXpp8SWffOKvs8aykzonQMMnHdknYbgodcdIpLmGSz+v3h41HfV+3iehWhYVB9bPZNRvP2+nYbq8XVb9i2knUmEZt12tMo8Ylqo347wSA9liTAwAAAACATShqgePCty9o7uic9n9rv+Yfm1f2WFaZwxnt+ek92vXILs3sn9HsgVml96Z1/vHz+uDNDwb0CWqi/DI/alK538nRMInhbs8RpmDQz88/qH52inm3fRnkPY8S037oNqZR2vUS037EpV/3gcIGsLFR5AAAAAAAIGbq62t0Wmcj7HHN2kVZm+PcyXPa9fAuzR2dU/ZYVg999SHNfWVOe768Rzsf3qmZfTPaPr9d05lpbZvbpuSWpF458UrX1xmkqMnOOCVJwxYAhj2NUuP14trPsOJ0z+uixnSc70Uc7wOAeGG6KgAAAAAAYqS+iHi9cLG+GLG+ONHpuMb39f3NCiON+5q1u7h4Uek9ac0emNXuL+1W5mhGM/tmVHWqKn1UUmIqIcMw5Hu+qk5Vnu3JtVzZD2x9/8nv6/Qfne5HeCKL01ocaG/U6yeMujhEUr+G7x6AsChyAAAAAAAQM61GWTRuD3tcP/bduHRDCycWlN6b1rb5bZrZN6MdCztkL9uqOlW5liun4Cg1naq90ikl00lN7ZzS26+9PbAiR5hEaNymqerm2uhPEaDbxatHWWiIQx/CGOQz2u8YjEtMAURDkQMAAAAAALSVv5ZXcmtSqemUkluTMpOmqk5V9rItz/YU+KujSiYMmQlTZtKUmTKVSCVqr6mElq4vKXMk01M/2iVVoyxeHcaok6Kjvn69D/tebr5o8zAS3a22R4lN1AXuB2WY8RyUQS/kHkWzOMbhuwRgMFiTAwAAAACAmMjn85961V25cmWk73PXcpIhGaahwA/kllyVPiqpkCvIumvJKTrybE++5yvwAylYne7KlIyJWpvcW7nuA9IH4zhNVZx/dV4vdtRfw1BfUDrqQtZh4znse95p8fQ4FzwG+YwO6nPHOZ4AesNIDgAAAAAAYuDFF1/UI488olOnTn1m3/pppEb1XoHku77coqvy/bISUwm5livDMOTZnioPKnJLrjzbU9Wt1goe1aBW9OiTVgnVTqMMxnGaqrgVOFotTD3oBatbFQJa3fNG3RY44hD3+ueLq2EUOHo9d7P2YZ8ZAOOHIgcAAAAAADHx7rvv6qmnnvrM9vWFj1G8zx7LSpKcoqPyclmTdyZlGIacgiNjorbYuFtyVXlQkVNy5FqfLnYYpqHs8Wy7j96TTknvdgnjdknVqO2iilOivZU4961RlHgO+56Pm2HFYBD3oZviGIDxQpEDAAAAAAC0NXd0Tq7lyl62Zd21lJhMyPd8paZTMhOmAj+QZ3tySo4qKxU5xdVCh+XJs2uvzOHe1uPY6MY5gR7HvsexT+OOmAKIK4ocAAAAAACgo4NPHNTNyzdrhY2J2sLjqemUzKQpBVLVrcq1XDnFWqGjPqrDXrF16MlDI+lzu2Rsu4Rt1HZRdXvOsCNXRp2MjtrPXqdrivL5h33P22n32XuNadRnZlgxiNN9ADA+WHgcAAAAAAB0tPjaolzLVSFfUHGpqEKuoMIHBRVytb9LH5Zk3bFUvldW+X5ZlZWK7BVb8qXTF04PtG9xXr+gk14Tt42ffdiLNkdd1DtsP5sdF2aKsbgnwjvFUxpeTDu1G/eYht0PYHwxkgMAAAAAAIRy9o2zOv/4eRVuF+RarpLppBKphGRKQTVQ1a3Ks7y1ERxBEOjZq8/27fqdkpRxT8I2Wv95Ov16v9m2eptmbTv9Ir/T9m7XKGklaj87tWuml3j2ImpMoybdBxXTTve825j28qxFFSam4/bvBIDOGMkBAAAAAABCyR7P6unXn1bxblHL7y/XRnPkCirmi2ujOUp3SirkC3KLrp65/IwyRwe/Fset5zZn4rJdMnuY1+p0vaj97OWacdeu/6OK6bgL89k3wucE8FmM5AAAAAAAAKHNPzav5yvP69J3Lumd772jxFRCgR9IkgzTkGd7OvTkodBTVIVJOg4zaT+odoM6V7fn6OWao2jbr3U1ehG1cNDreQfVflgx7ff9GNW/FQDijyIHAAAAAADo2plXz+jMq2e0dH1JubdykmojPTKHBz9yAwAAoI4iBwAAAAAAiCxzJKPMEQobAABgNChyAAAAAADGkmEYa++DIIh0XNR9649pd21sLt0sJM20OgAA9AcLjwMAAAAAxk69AFEvMKwvSIQ9rl6gqL/C7ms8NwAAAEaHkRwAAAAAgLFUL1y0KkJ0Oq6XERj1IkjYQkf+Wl65a6vrVhzLau7oXORrI74YnQEAwPBR5AAAAACANnqdEqmb9kx7FB/tCiLd3KuLixd149INJbcmJUPSajPXcnXwiYNafG2xn90GAADYdChyAAAAAEAL66c6MgyjZXK73XGdplPqtA/D0XhvW/0dtsBx++ptnTt5TundaS2cWFBqOiXDNOS7vpyiI3vZ1s3LN/XS5Es6+8ZZZY9nB/K5AAAANjqKHAAAAADQRq9TInUSdtoj3/X1b/7w3+ivfvBX+vD/+VCStPdn9+rhX3xYh3/1sMwESy5G1e0omsZ71dj+9tXbuvDtC5o7OqfZA7NK700ruTWpwA/kFl2Vl8uy7lpKTadUyBd0/vHzevr1p/v2eQAAADYTihwAAAAAMCJhk+vX/+C6Xv/11yVDSk4llZpOKfADvf+j9/VXP/wr/fDXf6i/+9t/Vz/7zM8OodfxEbZA1O64bgscjcc2a3/u5DnNHZ3T3NE57f7Sbm2b3yYzacotuSrfL2vyzqQSkwmZE7XCVOF2Qa+ceEX6r0J3AwAAAKsocgAAAADACIRNrv/x3/9jvX3hbe09tFfpvenatEcy5FU8VR5UVP64rPK9sv74P/lj5f8sr198+ReH0PvRWz81WP3vusbpwtodt/6/6/dHXaT84uJFpfekNXtgVru/tFuZoxnN7JtR1amq9FFJiamEDMOQ7/mqOlV5tifXcmU/sOVcdCSW6AAAAOgKRQ4AAAAAGJFO0x79i2f+hf7if/0LLZxY0OzDs5qem1ZiS0K+66vyoCLrnqXJO5NKTiVlJkz92R/8mVzLlR4a9icZjVaFh8btYY/rdn+zY25cuqGFEwtK701r2/w2zeyb0Y6FHbKXbVWdqlzLlVNwlJpO1V7plJLppKZ2Tsm54XS8HgAAAD6NIgcAAAAAtNGPKZGa6TTt0e2f3NafX/pzff7rn9feQ3u15+AebX9ouwzTqBU47lhKbEmsrcXhV31V3are/p/flv5DSaxjPXT5a3klt9amE0tuTcpMmqo6VdnLtjzbU+CvjiqZMGQmTJlJU2bKVCKVUCKVkBKSscQi9AAAAN1gZToAAAAAaKFedGg31VGY49ZPixS2CPKDf/QD7VjYoZl9M9r18C7t/bf2av6xee16ZJe2Zbdp656t2rJri6Z2Tmlyx2TttX1SyXRS+t97+NBj4NFHH1U+n1971a3f1rj9ypUra38P6n3uWk4yJMM0aouMl1yVPiqpkCvIumvJKTrybE++5yvwAylYfWZM1QofhinleokMAADA5sNIDgAAAABoY9BTIjU7rnyvrA/f/lALX1/QltkttdfOLUpuTapaqSqxJaHklmTtNZX85P2W2igC628tyQ75AcfMCy+8oFwu1zSu7e7B+n0DfR9IvuvLLdYWGU9MJeRargzDkGfX1lFxS64821PVrdYKHtWgVvQAAABA1yhyAAAAAEDM5K7lNJGcUGJLQhPJCQV+IHvFljFhyHd9VSvVWlJ8ddTA2vRHq1MgGROGgtzGTZpns83n4mq3ff2+U6dODeR9fntt9IhTdFReLmvyzqQMw5BTcGr3zvPlllxVHlTklBy5VkOxIwgUZDfufQMAABgEpqsCAAAAgAaHDx/WL/3SL+ntt99ee9X9+Mc/Xnv/8ssvD+T93f/vrswJUwokr1L79X/pw9q0R6U7JVVWKnIsR17lk6mP1k+ZNWFMyLjD2g7DNnd0Tq7lyl62Zd21VFwqqpArqJArqJgvqvRhSdZdS+WPy7V7WFwtdFhebc0OL5Ayo/4UAAAA44WRHAAAAADQ4Jd/+ZflOE7TfceOHVt7/8wzzwzkff5P86p6VXm2J6fgyLpryUyYckqODNNQtVKVU3TWkuTVSlW+68uv1l5VvyrtiPjh0ZODTxzUzcs3lZpOyZyoLTyemk7JTNaKVlW3Ktdy5RQdVVYqa6M67BVbOjjq3gMAAIwfihwAAAAA0EQqlWq6fXp6eu39zMzMQN4bX6ktXF15UFH5frm2mHggOQVHZsKUX/VrifKCU3uVHLllV1W7Kt+pTXuk5jM3YcAWX1vUS5MvqZAvSJI821MqnZKZMmUYtSmrPNuTa9WmraqsVGoFDl/SoiRmqwIAAOgK01UBAAAAQMxs//x2bctsk33PlnXHUmmptDbtUSFfUHGpKOuOpfL9suxluzYaYHVUh1N2pG2Sto/6U2xeZ984K7foqnC7ds8e5B6omCuu3bvSndLa/bPuWwqCQM9efXbU3QYAABhLjOQAAAAAgJgxTENf+y++psu/cVnJ6aRkSp6zOiIgWfutmu/68sqenNK6aY+KjjzLk35BChgSMDLZ41k9/frTeuXEK7If2JraOaVEKlFbEN4P1kZz1EdwPHv1WWWOZqT/bdQ9BwAAGD8UOQAAAAAgho79x8d0/Q+u695f3lMQBGvTHk0kJyRDtbU3Kp9e38G6a2nvz+7V7SO3mfZoxOYfm9fzled16TuX9M733lFiKqHAX10c3jTk2Z4OPXlIpy+cHnFPAQAAxhtFDgAAAACIqV9789f0Oz/zO/r4vY81bU3XihyTEzJMQ0E1UNVdXZy85Kh8v6xdD+/Ssz95Vi+++OKou45VZ149ozOvntHS9SXl3spJqo30yBzOjLhnAAAAGwNFDgAAAACIK0P6B//vP9DFJy7qr//lX6v0UUnJLUlNTE5IgeTarryyJzNl6ku//CWdefXMqHuMFjJHMsocobABAADQbxQ5AAAAACDmnrj4hG79H7f0k//+J/rgzQ9UyBUkSem9aS383IK+9htf08LXF0bcS/STYRhr74Og9dxjrY7rpn2z/WHbAwAAjBpFDgAAAAAYA/t+fp/2/fw+SbVFxyWtLUKOjaVeYAiCQIZhdCxENDuu/t/1xYpW7Zttp7ABAADGBUUOAAAAABgzFDc2vvXFinaFirDHNaoXMhrbRClw5K/llbu2ut7Isazmjs41PW7fy12dds2t5zq3vfVc631R2/ZyzW60uk7Y8zdrH6ZtY7thX29Q1+w1nu3O1+kc3ca012dsWPei38/ooOPS7nz9+t4CiBeKHAAAAAAAbCKdChlhp6q6uHhRNy7dUHJrUjIkrR7qWq4OPnFQi68t9qvLHe17eTyTl+2SufV9UZLP7eIRtdjUqa/97meUa/Yaz26uH+W4fup3TFu16yWmo4hLHPsAYPAocgAAAAAAsEmEGanRuLZH4/G3r97WuZPnlN6d1sKJBaWmUzJMQ77ryyk6spdt3bx8Uy9NvqSzb5xV9nhWUudEaJjkc7skbKfkbtQiyKCLJ53iEib5vH5/t/HoNgncqrAwqH42u2bjeTsd2+31oupXTDuJGtOo7XqN6bDiAmDzYowzAAAAAACbSH39jvqIjW6mubp99bYufPuC5o7Oaf+39mv+sXllj2WVOZzRnp/eo12P7NLM/hnNHphVem9a5x8/rw/e/GBQH0XSeCdC+1GUaTwuTMEgalK9mwR4p+PCTlnU6rhu+9LPz9zs3KN4DruNaZR2vcR0VHGpizJ6B8B4osgBAAAAAEDMhC1AdFuoCILgU6/6trDOnTynXQ/v0tzROWWPZfXQVx/S3FfmtOfLe7Tz4Z2a2Tej7fPbNZ2Z1ra5bUpuSeqVE6+EPj/CCVsAGPZUPY3Xi2s/w4pjcjxqTMf9XkQVx3sIoP+YrgoAAAAAgBipLwheL1y0mj6q03GN78MUMxoXI1/f5uLiRaX3pDV7YFa7v7RbmaMZzeybUdWpqvRRSYmphAzDkO/5qjpVebYn13JlP7D1/Se/r9N/dDpKOPqmXwttkzQdvFH/An/UxSGesZpe4rLRijUA2qPIAQAAAABAzLQqSDRuD3tcN9dp1fbGpRtaOLGg9N60ts1v08y+Ge1Y2CF72VbVqcq1XDkFR6npVO2VTimZTmpq55Tefu3tgRU5el1Ie5DTGPWCJO0n+lEE6HYRcaZZ6mxUxaBepzoDsPFQ5AAAAAAAAG3lr+WV3JpUajql5NakzKSpqlOVvWzLsz0F/uqokglDZsKUmTRlpkwlUonaayqhpetLyhzJ9NSPdknVbtYOCJucjdqun+KQqL31XO2zN1tgepAxaXXuXpLYURe4H5Q4PGO9GkS8eo1LHL43AIaHNTkAAAAAAIiJRx99VPl8fu1V9/HHH+vKlStrfw/7fe5aTjIkwzQU+IHckqvSRyUVcgVZdy05RUee7cn3fAV+IAWr02SZkjFRa5N7KxcpJr3qtEB1q+Rp1Hb9EOdfoteLHfXXMNQXsI66kHW3IwCGZZTPWK8G+Yz2Epc4xwzA4FDkAAAAAAAgBl544QWdPHnyMwuD163/eyTvA8l3fblFV+X7ZRWXiirkCip9WFL5XlmVBxW5JVee7anqVmsFj2pQK3r0SWOye33Se1i/9B7WNFVxK3C0KjBELTx0c91W28Lc83Gc4igOfWhnVLEap3sIYLiYrgoAAAAAgJjIZrNNt+/cuVOnTp1a+3vY7/Pba6NKnKKj8nJZk3cmZRiGnIIjY6K22LhbclV5UJFTcuRany52GKah7PHmn60f2k2nNE7GIUkb5741ihLPMKMExikG/TYOMeAeApsPRQ4AAAAAANDW3NE5uZYre9mWdddSYjIh3/OVmk7JTJgK/ECe7ckpOaqsVOQUVwsdlifPrr0yh3tbj2OjG+fkaxz7Hsc+jTtiCiCuKHIAAAAAAICODj5xUDcv36wVNiZqC4+nplMyk6YUSFW3Ktdy5RRrhY76qA57xdahJw+NuvufEXXu/kHM+d9t8rjTyJW4JKOj9rPeLqoon7/dscOOZ7vP3mtMoz4zcXimOj0TcbqHAIaLNTkAAAAAAEBHi68tyrVcFfKFtfU4Ch8UVMjV/i59WJJ1x1L5Xlnl+2VVViqyV2zJl05fOD3QvrVKfobZ3i7h2227KHpNvjb2dVALL3eKSbeLeoftZ7Pj2l1zXJLZ/XjG+hXTTu2GGdNhfvcAbByM5AAAAAAAAKGcfeOszj9+XoXbBbmWq2Q6qUQqIZlSUA1UdavyLG9tBEcQBHr26rN9u36UX3LHacRGu2t0+vV+s231Ns3advpFfqft/Ypl1H52atdML/HsRdSYRn3GBhXTTve825gOOy4ANi9GcgAAAAAAgFCyx7N6+vWnVbxb1PL7y7XRHLmCivni2miO0p2SCvmC3KKrZy4/o8zRwa/Fceu51oWAbtv00m7YovR/ENfqdL2o/ezlmnHX6zM2iJjGwbh89wDECyM5AAAAAABAaPOPzev5yvO69J1Leud77ygxlVDgB5IkwzTk2Z4OPXko9BRVYZKWvSQ2o7YdRjK1H9fo9hyjiGUvbfu1rkYvohYOej3voNoPK6Zx+u5RHAE2NoocAAAAAACga2dePaMzr57R0vUl5d7KSaqN9MgcHvzIDQAAgDqKHAAAAAAAILLMkYwyRyhsAACA0aDIAQAAAAAA0AfdLJjM9DkAAPQHC48DAAAAAAAAAICxxEgOAAAAAACAPmB0BgAAw8dIDgAAAAAAAAAAMJYocgAAAAAAAAAAgLFEkQMAAAAAAAAAAIwlihwAAAAAAAAAAGAsUeQAAAAAAAAAAABjiSIHAAAAAAAAAAAYSxQ5AAAAAAAAAADAWKLIAQAAAAAAAAAAxhJFDgAAAAAAAAAAMJYocgAAAAAAAAAAgLFEkQMAAAAAAAAAAIwlihwAAAAAAAAAAGAsUeQAAAAAAAAAAABjiSIHAAAAAAAAAAAYSxQ5AAAAAAAAAADAWKLIAQAAAAAAAAAAxhJFDgAAAAAAAAAAMJYocgAAAAAAAAAAgLGUGHUHAAAAAACDYxjG2vsgCCIdN4h9AAAAQD8wkgMAAAAANqh6kaFeYFhfdAh7nGEYCoJg7dWPfQAAAEC/MJIDAAAAADaweuGiU6Gh1XHtRmD0e3RG/lpeuWs5SVL2WFZzR+eaHrfv5Wjnv/Vc57a3nmu9r1Xbdm06tQ/bNur5u7lG1P41thv29QZ1zV7j2e58nc7RbUx7ea5btR/Evej3Mxo1Lt3cw0F/bwEAvaPIAQAAAACIrB9TUl1cvKgbl24ouTUpGZJWT+Narg4+cVCLry32oafh7Hu5eQKzXRK5vq/bZHm763Wrl/61a9+uf1GLTb3EI0o/o1yz13h2c/0ox/VTv2M67O9Qp3NGuV4/2gMAhociBwAAAAAglPoUVOut/3v9/jBTVN2+elvnTp5TendaCycWlJpOyTAN+a4vp+jIXrZ18/JNvTT5ks6+cVbZ41lJnROhYRKP7ZKw7ZK7Uds1axu2XVid4hIm+bx+f7efq9tEdLfx6LWfza7ZeN5Ox3Z7vaj6FdNOosY0arteYxolLlG+s51G3QzjGQAAhMeaHAAAAACAjpoVODpZvyZHo9tXb+vCty9o7uic9n9rv+Yfm1f2WFaZwxnt+ek92vXILs3sn9HsgVml96Z1/vHz+uDND/r1cZrqlFztpvBR167w0s/kaDfFlbDHhSkYRE2q9xKPbvvZqfjVbV/6+ZmbnXsUSfNuYxqlXS8xjRKXqN/ZMMdR2ACAeKHIAQAAAAAbWH00RadRFe2Oa1XgCLuYeLP2506e066Hd2nu6Jyyx7J66KsPae4rc9rz5T3a+fBOzeyb0fb57ZrOTGvb3DYltyT1yolXQl1vHI1imqL11+2UzB12/xqvF9d+hhXHpHjUmI77vQAAbDxMVwUAAAAAG1R9yqh6MaLT1FKtjlv/3/X72y1S3m6tjouLF5Xek9bsgVnt/tJuZY5mNLNvRlWnqtJHJSWmEjIMQ77nq+pU5dmeXMuV/cDW95/8vk7/0eneAoNNY9RrJ4y6OBTH4goAAP3GSA4AAAAA2MBaTRnV7O9WxzW+um3X6MalG9r58E6l96a1bX6bZvbNaMfCDm3dvVVTM1Oa3D6p1HTqk1c6pWQ6qamdU3r7tbd7CUdbvS6kHdeEMr+o/8S+lz/9inqObo4b5XPRy+ccplEVg6KORonDvQUAfIKRHAAAAACAoclfyyu5NanUdErJrUmZSVNVpyp72ZZnewr81VElE4bMhCkzacpMmUqkErXXVEJL15eUOZLpqR/tkqph13gI0+bWc58kmlsttD0scUjIjioeg0hWR13gflBG/Xz1w6Di1c13dv3+dkWiOHyfAAA1jOQAAAAAgA3o0UcfVT6fX3vVrd9W31spxUQAACAASURBVH7lypW1/YN+/+NXfywZkmEaCvxAbslV6aOSCrmCrLuWnKIjz/bke74CP5CC1amvTMmYqLXJvZWLGpaBCZNQ7scogm7F+Rfno4hHfTHqqAt8h41nHApY67fFueAxqmc0zjEBAHSHkRwAAAAAsMG88MILyuVyTaeK6rRt0O9rGyTf9eUWXZXvl5WYSsi1XBmGIc/2VHlQkVty5dmeqm61VvCoBrWiR5+0Sqi2GmXQqW27dq2mvhn0As1xLXCMKh6t7k2Ye76+X/06bhjqny+uhhWrbr+z9f1R2wIAhosiBwAAAABsQNlsNvT2U6dODe/9U6f03u++J6foqLxc1uSdSRmGIafgyJioLTbullxVHlTklBy51qeLHYZpKHu8+Wfrh26S3t22G2ZCNE6J9lbi3LdGUeLZrrgwDvdn0EYdgzBTp0WZhg4AMHwUOQAAAAAAQzN3dE6u5cpetmXdtZSYTMj3fKWmUzITpgI/kGd7ckqOKisVOcXVQoflybNrr8zh3tbjiJNBJHpHnTzuRRz7Hsc+jTtiCgDoJ9bkAAAAAAAM1cEnDtbW4LhjqbhUVCFXqL3yBRWXiirdKcm6a6n8cVmVlcraqA57xdahJw+Nuvux1m3yuNMUUXFJRkftZ6/9jvL5G9f9aLYGSNQ1QbrVbjRJrzGN+szE5ZkCAGwcFDkAAAAAAEO1+NqiXMtdK2oUcgUVPqgVOopLRZU+LMm6Y6l8r6zy/Vqhw16xJV86feH0QPvWKXHbbbsw5+xXsrfX8zX2c9BrhbTa3u2i3mH72ey4TusuhOnPqIV5ZocV07Dfk1EXeMLsb3dMnNc5AYDNiOmqAAAAAABDd/aNszr/+HkVbhfkWq6S6aQSqYRkSkE1UNWtyrO8tREcQRDo2avP9u36nZKU7ZLe3bYL27YX688f5tf7jdvqbZq17fYztVpQPEzbdqL2s1O7ZnqJZy+ixjTq8zWomHa6593GdJBxifKdaNcWADB8jOQAAAAAAAxd9nhWT7/+tIp3i1p+f3ltyqpivrg2mqN0p6RCviC36OqZy88oc3Twa3G0mkaoUzKz3fRD7bbHJUk6zMXSe4lH1H6Owz2Iql3/RxXTOOjlO9tp/0Z4bgBgI2EkBwAAAABgJOYfm9fzled16TuX9M733lFiKqHADyRJhmnIsz0devJQ6CmqwiQde0lMjqrtsM7f7TnGLZbdrqsxCFELB72ed1DthxXTUcWlX+cAAAwWRQ4AAAAAwEidefWMzrx6RkvXl5R7KyepNtIjc3jwIzcAAAAw3ihyAAAAAABiIXMko8wRChsAAAAIjyIHAAAAAAAx081C0kynAwAANjMWHgcAAAAAAAAAAGOJkRwAAAAAAMQMozMAAADCYSQHAAAAAAAAAAAYSxQ5AAAAAAAAAADAWKLIAQAAAAAAAAAAxhJFDgAAAAAAAAAAMJYocgAAAAAAAAAAgLFEkQMAAAAAAAAAAIwlihwAAAAAAAAAAGAsUeQAAAAAAAAAAABjiSIHAAAAAAAAAAAYSxQ5AAAAAAAAAADAWKLIAQAAAAAAAAAAxhJFDgAAAAAAAAAAMJYocgAAAAAAAAAAgLFEkQMAAAAAAAAAAIwlihwAAAAAAAAAAGAsUeQAAAAAAAAAAABjiSIHAAAAAAAAAAAYSxQ5AAAAAAAAAADAWEqMugMAAADYfAzDWHsfBEHXx63f3uwcYc8PAAAAABhvjOQAAADAUNULEPXiQ2PBIuxxQRCsvRrbrd/X6vwAAAAAgPHHSA4AAAAMXb0w0akIEfa4XuWv5ZW7lpMkZY9lNXd0rulx+16Odv5bz3Vue+u51vtatW3XJsy5orTvdM71wp6/WfswbRvbDft6g7pmP+934/k6naPbmPbyXLdqP4h70e9nNGpcon62sG0BAAA2I4ocAAAAGEv9mJLq4uJF3bh0Q8mtScmQtHoa13J18ImDWnxtsQ89DWffy82TmO2SyPV9vSTae9Vr/1q1bxWPTtdsp1Nf+93PKNfs5/3udL4ox/VTv2M6iO9QlLgM6npR7j8AAMBmQJEDAAAAY6fVFFX1fZ1Gfdy+elvnTp5TendaCycWlJpOyTAN+a4vp+jIXrZ18/JNvTT5ks6+cVbZ41lJnROTYZKP7ZKw7ZK7UdsNQ6e4hEk+r98f9nPV93WbiG5VWBhUP5tds/G8nY7t9npR9SumnUSNadR2vcY0Slx6+c7G+fsOAAAQN6zJAQAAgA2n1XodUq3AceHbFzR3dE77v7Vf84/NK3ssq8zhjPb89B7temSXZvbPaPbArNJ70zr/+Hl98OYHA+1vp2RnN4WPVgb1K/BuE7VhjgtTMIiaVO8mAd7puLBTFnVz//oRz7DXbzz3KBLn3cY0SrteYholLr18Z/v1fQcAANhMKHIAAABg6OojLTqNuAh7XLv2jYWOcyfPadfDuzR3dE7ZY1k99NWHNPeVOe358h7tfHinZvbNaPv8dk1nprVtbpuSW5J65cQrka4fN3FKkoYtAAx7GqXG68W1n2HF6Z7XRY3puN8LAAAADAbTVQEAAGCo6tNJ1QsX64sQzaadanVc4zmb7WsscFxcvKj0nrRmD8xq95d2K3M0o5l9M6o6VZU+KikxlZBhGPI9X1WnKs/25Fqu7Ae2vv/k93X6j073KQrDRdJ3+Ea9fsKoi0NxLK4AAABgY6LIAQAAgKFrtVB44/awx4Xdd+PSDS2cWFB6b1rb5rdpZt+MdizskL1sq+pU5VqunIKj1HSq9kqnlEwnNbVzSm+/9vbAihy9LqQdZpHmUSSdKa58oh9FgG4XER9loSEOfQhjVMWgXu5/3GMKAAAwbBQ5AAAAsCnkr+WV3JpUajql5NakzKSpqlOVvWzLsz0F/urIkQlDZsKUmTRlpkwlUonaayqhpetLyhzJ9NSPdknVsGs8dNMm7DGDNOrr1/uw7+XmizYPMtHd6ty9JKzjds+HGc9BGVS8on5ne20LAACwmVDkAAAAwNDk8/lP/T03Nzew7e+++65OnjwpSbpy5Yqm/2JaMiTDNBT4gdySq9JHJVWdqgI/kFN05NmefM9X4AdSsDr1lSkZE7U2ubdyPRc5+q1Z0n79vlGJ86/ORxGXXgsBYeM57M/WavH0ej/aPZ+jNqpntJeYxDmeAAAAo0KRAwAAAEPx4osv6pFHHtGpU6c+s6/baanCbG/6PpB815dbdFW+X1ZiKiHXcmUYhjzbU+VBRW7JlWd7qrrVWsGjGtSKHn3SriDRKYHZbF+n0QmjnKYqbsnYVgtTD3rB6naFgDBJ624LHHGI+/pCRxwNK1bdfGf72RYAAGAzocgBAACAoXn33Xf11FNPfWZ7Npttenwv29e/P3XqlPLba6M8nKKj8nJZk3cmZRiGnIIjY6K22LhbclV5UJFTcuRany52GKah7PHm1+2HbpLe3bRrl2geRKI3Ton2VuLct0ZR4jnsez5uRh2DqN/1XtsCAABsVBQ5AAAAsCnMHZ2Ta7myl21Zdy0lJhPyPV+p6ZTMhKnAD+TZnpySo8pKRU5xtdBhefLs2itzOF5TVcXNqJPHvYhj3+PYp3FHTAEAADYeihwAAADYNA4+cVA3L9+sFTYmaguPp6ZTMpOmFEhVtyrXcuUUa4WO+qgOe8XWoScPjbr7XWmXxI3DCI6wI1BGnYyO2s9ep2uK8vmHfc/baffZe41p1GcmLs8UAAAA+sscdQcAAACAYVl8bVGu5aqQL6i4VFQhV1Dhg4IKudrfpQ9Lsu5YKt8rq3y/rMpKRfaKLfnS6QunB9q3VknhTonyOKx70GvyuPEzDOozdYpxt4t6h+1ns+PaXXNckvFhntlhxTTs92QYMe3lOzsO33cAAIC4YSQHAAAANpWzb5zV+cfPq3C7INdylUwnlUglJFMKqoGqblWe5a2N4AiCQM9efbZv1++UpOx2XY127YZhfd86/Xq/2bZ6m2ZtO/0iv9P2qLFs1o8o/ezUrple4tmLqDGNmnQfVEw73fNuYzrIuHT7fIdpCwAAsBkxkgMAAACbSvZ4Vk+//rSKd4tafn+5NpojV1AxX1wbzVG6U1IhX5BbdPXM5WeUOTr4tThuPde6EBCl3bhol8we5rXCxLmb7f24Zty16/+oYhoHvXxnN/r3HQAAYBAYyQEAAIBNZ/6xeT1feV6XvnNJ73zvHSWmEgr8QJJkmIY829OhJw+FnqIqTNKxl8Rkv5Oa/TxfP87V7TlGFcuobfu1rkYvohYOej3voNoPK6ajiAtFDAAAgO5Q5AAAAMCmdebVMzrz6hktXV9S7q2cpNpIj8zhwY/cAAAAAAD0jiIHAAAANr3MkYwyRyhsAAAAAMC4ocgBAAAAYE03C0kzrQ4AAACAUWPhcQAAAAAAAAAAMJYYyQEAAABgDaMzAAAAAIwTRnIAAAAAAAAAAICxRJEDAAAAAAAAAACMJYocAAAAAAAAAABgLFHkAAAAAAAAAAAAY4kiBwAAAAAAAAAAGEsUOQAAAAAAAAAAwFiiyAEAAAAAAAAAAMZSYtQdAAAAQHOGYay9D4Kg6+M6tQ/TLsz1AQAAAAAYFUZyAAAAxFC90FAvLjQrPHQ6LgiClsUJwzDW9gdB0LRdu/YAAAAAAMQBRQ4AAICYqhcYOhUawh7XrE0n9WIIAAAAAABxxHRVAAAAm1TY6bDa+fjmx/rX/+xf670fvaf7N+9LknY9vEv7/939euwfPaaZL8z0pa8AAAAAADRDkQMAAGCTalyHo7HQ0WkUx+X//LL+7Pf/TAqkqZkp7fzCTvlVX9aHlq79j9d0/ZXrOvb3j+mbv/nNgX0GAAAAAMDmRpEDAAAAXfvnv/DPlb+WV+ZwRtN7p5VKpyRJru2q8qAi+74t666lf/U//Ct9eP1DPfWDp0bcYwAAAADARkSRAwAAIKbqIylaLTre7XHN2kRx/vHzWvq/l7Tv1D7NPjKr6blpJaYSqjpVVVYqsu5ZsrZZmpiakJEwdOuNW7rw712Qjke6HAAAAAAALVHkAAAAiKF60aJeuGg1tVSn4xrfN7Zbf731WhVB/vzSn+v2/3VbCycW9Lmf+Zz2Htqr7Q9tlwzJXrZV+qikxFRChmko8AP5rq+qU9X7P35f2i7pQA9BAQAAAACgAUUOAACAmGo10qJxe9jjutnfat+P/psfaecXdmrHwg7t+qld2vMzezR7YFble2UVcgUFfqCqU5VbduVarpySI6foqJKuyP0TlyIHAAAAAKCvzFF3AAAAAOOhmC/q/s372vq5rdqyc4smZybX1uIwE6YmJieUmEp89rUloWQ6Kd2TjFL4KbUAAAAAAOiEkRwAAAAIJXctt1bMMCYM+Z4ve9mWDMn3fHm2J9/zpaA23ZVhGjInTJmJ2suYMBTkoq0DAgAAAABAM4zkAAAAiJErV67o2rVrn3rVXbx4ce39iy++OPT3y3+9LHPCrE1JVaktMl5cKqqQK8j6yFJluSK35MqzPVXdqoJqoMAP1ooeE8aEdL/7mIyb+hopYRaMb3bc+u3tztFsX9hrAwAAAMBGwUgOAACAGPnxj3+sIAj03e9+9zP7nnjiibX3L7zwwtDfz3xxpjZio+yp8qAi664lc8KUU3RkTpiqOlU5xdoaHK71SbHD93z5nq+qX5V2hQ7FWFq/wHu92NBsfZNOx3VaT6VVgaPVAvUAAAAAsFFR5AAAAEAo2a9k5Vd9VR5UVL5fVnJrUoEfKFVIyUyYCqqB3LIrp+DUXiVHnuXVih1OVYEfKMhu/KR7vbBQL2D0elyjevGisU2Ugkb+Wl65azlJUvZYVnNH55oet+/lrk8tSbr1XOe2t57rfJ7Gc4Rp06ptt+27PXc354/at6jx6Nf1BnXNXuPZ7nydztFtTHt9rod1L/r9jA6yXT/+rQAAAJsTRQ4AAACEMj03rdmHZ1W+W1ZpR0lmwlTVrSo1ndJEcqI2jZVTlVNy5DxYfa2O6nDLrjQrKT3qTzEe1hcwBjE64+LiRd24dEPJrUnJkLR6StdydfCJg1p8bbHna4S17+Xuk95hzxvlmv06vxQt+TyIePQSiyj9jHLNXuPZzfWjHNdP/Y5pq3a9xHQQzxoAAMCgUOQAAABAaN/4zW/o0lOXlNyWlGEY8ireWpFDknzXl2u7coqOKg8qsh/YcoqOvLIn/cqIOz8mGosY60duhC1wtDr29tXbOnfynNK701o4saDUdEqGach3fTlFR/ayrZuXb+qlyZd09o2zyh7PSuqcCA2TfG6XhO2U3K3vi5JA7Tb52+u5158/TPJ5/f5Bx6NVYWFQ/Wx2zcbzdjq22+tF1a+YdhI1plHb9RrTXuPSr+8sAABAOyw8DgAAgNAe/fcf1RdOfkErt1ZUyBdUyBVU+GD1v/mCih8WZd2xVL5Xlv2xrcpyRdY9S1/81helR0bd+/HXuLB4mLU56m5fva0L376guaNz2v+t/Zp/bF7ZY1llDme056f3aNcjuzSzf0azB2aV3pvW+cfP64M3Pxjo5wmTII+S8OxUfOlHEjVMYrjbcwwiHu1iMah+Rol/P+IZ9vqN5x5FUr3bmEZp10tMo8ZlVPEEAACbG0UOAAAAdOWpP35KC19f0MrfrKiYK9YKHLmCivmiih8WVfqwpNKdkqy7lkp3S/qpb/6Unvxfnhx1t4emXQEiynF1QRB86lXf1njOVqM9zp08p10P79Lc0Tllj2X10Fcf0txX5rTny3u08+Gdmtk3o+3z2zWdmda2uW1KbknqlROvhOobwgtbABj2tD+N14trP8OKY6I9akzH/V4AAAAMGtNVAQAAoGtP/+Bp/ck//hO99TtvqfhhUROTE0pM1v6npWd7qlaqmtgyoZ/7jZ/TqRdPjbazQ1SfVqpeuGi1nkan4xrPGUazokm97cXFi0rvSWv2wKx2f2m3Mkczmtk3o6pTVemjkhJTCRmGId/zVXWq8mxPruXKfmDr+09+X6f/6HS3oRiZKOs3oH9GHf9RF4d47npHTAEAQLcocgAAACCSb/6Tb+rYf3RMb/7Om3r/T97Xvb+8J0nadWCXvvjNL+qxf/iYtj+0fcS9HL5WRYnG7WGPC3uddu1uXLqhhRMLSu9Na9v8Ns3sm9GOhR2yl21Vnapcy5VTcJSaTtVe6ZSS6aSmdk7p7dfeHliRYxgJ6VEkTPlF/Sf6Ef9uFxEfZVI8Dn0IY5ye0XGJKQAAGB2KHAAAAIhsx74d+oX/9hdG3Q20kb+WV3JrUqnplJJbkzKTpqpOVfayLc/2FPiro0omDJkJU2bSlJkylUglaq+phJauLylzJNNTP9olVQeRvGx1vWEmTOOQlL31XO0zN1tgepCJ7kHEP+oC94MyzHgOShye0fU2QkwBAMDwUeQAAAAA+iCfz3/q77m5ub5uv3fvnt555x2dPHlSknTlypVQ76f/YloyJMM0FPiB3JKr0kclVZ2qAj+QU3Tk2Z58z1fgB1KwOuWVKRkTtTa5t3I9FzlGZdhJ0zj/6nwUCeNe4x82nsP+bK0WT6/3o1lRKS7i+oyOc0wBAMBosfA4AAAA0KMXX3xRP/rRjz6zMLjUfMHwXrav/zv0+0DyXV9u0VX5flnFpdqC8aUPSyrfK6vyoCK35NbWU3GrtYJHNagVPfrk1nPNX9LgEtStkqaDumack8etYjHIvvYa/24LHHGIexz60E6cYhXWOPUVAACMBiM5AAAAgD5499139dRTT31mezabbXp8t9t3796tU6dOrf0d9n1+e21kiFN0VF4ua/LOpAzDkFNwZEzUFht3S64qDypySo5c69PFDsM0lD3evE/90G46pXEyDsnjOPetUZR4tiucjMP9GTRiAAAANiqKHAAAAMAGNnd0Tq7lyl62Zd21lJhMyPd8paZTMhOmAj+QZ3tySo4qKxU5xdVCh+XJs2uvzOHxnKpqWMY5eRzHvsexT+OOmAIAgI2M6aoAAACADe7gEwdVyBVk3bHWpqoq5Aoq5AsqLhVVulOSdddS+eOyKiuVtVEd9oqtQ08eGnX3uzLsJG63yeNO0zXFJRkdtZ+99jvK5281Fdr6cwx6aq66dqNJeo1p1GcmLs9UVCw+DgAAOqHIAQAAAGxwi68tyrXctaJGIVdQ4YNaoaO4VFTpw5KsO5bK98oq368VOuwVW/Kl0xdOD7Rvg0xgNjt3PxO+vZ6rsX+DikWvBZWo/ew2/uOSjO8UT2l4Me3UbjPFFAAAbF5MVwUAAABsAmffOKvzj59X4XZBruUqmU4qkUpIphRUA1XdqjzLWxvBEQSBnr36bN+u3ykZ2y7p3Wl7s1++148ZRuEgzK/3G7e161+nX+R32t5NLNuJ2s8o8e8lnr2IGtOoz9WgYtrpnncb037HZVDPKAAAgMRIDgAAAGBTyB7P6unXn1bxblHL7y+vTVlVzBfXRnOU7pRUyBfkFl09c/kZZY4Ofi2OQU0j1C5ZHIdfhLfr3zCv1el6UfsZ9/j3ol3/RxXTcddrTAEAwObGSA4AAABgk5h/bF7PV57Xpe9c0jvfe0eJqYQCP5AkGaYhz/Z06MlDoaeoCpN47CU52Wtic5CJ0X6cu9tzjCqWUdt2u67GIEQtHPR63kG1H1ZMRxEXChkAACAqihwAAADAJnPm1TM68+oZLV1fUu6tnKTaSI/M4cGP3AAAAACAfqLIAQAAAGxSmSMZZY5Q2AAAAAAwvihyAAAAYOgMw1h7HwRB18e1a79+X7t2na6NzaWbRY+ZVgcAAACIDxYeBwAAwFDVCw31AkNj4SHMcUEQtC1Q1Pc3O6bdPgAAAADAeGEkBwAAAIauXmAIgqBlkaOb4wYpfy2v3LXVdSuOZTV3dG4k/cBgMToDAAAAGE8UOQAAALDhhJ3Oqt1ojouLF3Xj0g0ltyYlQ9Lqoa7l6uATB7X42mJf+wwAAAAA6B5FDgAAAGwozYoa60eEtNpXd/vqbZ07eU7p3WktnFhQajolwzTku76coiN72dbNyzf10uRLOvvGWWWPZwf7gQAAAAAALVHkAAAAAFbdvnpbF759QXNH5zR7YFbpvWkltyYV+IHcoqvyclnWXUup6ZQK+YLOP35eT7/+9Ki7DQAAAACbFkUOAADw/7N39zFunAee539VTbLbYrfUeotItrcVZfyWKEqsjuTkZleRdmLA98dl70aSkdhejAdyMNjB3UFY4PafuTE83vXN4u5wAxi3twvs4qwMotiJtfLiFjhMACHnaNejyWasGBMbWkwmWlsTmWxZL2mJb8WqYtX9wWanTfOlWCySxe7vByDErqqnnocPqw34+fXzPMDINWdQ9NpnI+h1UTlz9IyyS1lll7La9cguzS3MyUyacsqOqneqmr45rcR0QuaUKUkqXi/qlSOvSH8wkuYBAAAAAFoQcgAAAGCkmqFFM7hYv1xU69JS3a5rfd883xqIdCrXeu7cyXNK705r50M7teuRXcosZTS/d151u67yR2UlZhIyDEOe66lu1+VarpyKI+ueJfucLbFFBwAAAACMHCEHAAAARq7Tht+tx4NeF8W5K+evaPHIotJ70ppbmNP83nltW9wma8VS3a7LqTiyi7ZSs6nGK51SMp3UzPYZ2VfsjvcFAAAAAAyPOe4GAAAAAONWuFxQcktSqdmUkluSMpOm6nZd1ool13Lle6uzSqYMmQlTZtKUmTKVSCWUSCWkhGQsj2ZJLQAAAADArxFyAAAAYGQefvhhFQqFtVdToVDQpUuX1n6+ePHiSN/nL+clQzJMo7HJeNlR+aOyivmiKrcqsku2XMuV53ryPV/yV5e+MtUIPgxTyofqEgAAAADAAFiuCgAAACPxwgsvKJ/Pt10yyvf9jx0fy3tf8hxPTqmxyXhiJiGn4sgwDLmWq9q9mpyyI9dyVXfqjcCj7jdCDwAAAADAWBByAAAAYGRyuVzH4+vPHTt2bKTvC1sbs0rskq3qSlXTN6dlGIbsoi1jqrHZuFN2VLtXk1225VRawg7fl58j7AAAAACAUSPkAAAAwKaXXcrKqTiyVixVblWUmE7Icz2lZlMyE6Z8z5drubLLtmp3a7JLq0FHxW3s2eH6UmbcnwIAAAAANh9CDgAAAEDS/if36+qFq41gY6qx8XhqNiUzaUq+VHfqciqO7FIj6GjO6rDuWtLnx916AAAAANicCDkAAAAASSdfP6mXpl9SsVCUJLmWq1Q6JTNlyjAaS1a5liun0li2qna31gg4PEknJLFaFQAAAACMnDnuBgAAAABxceqtU3JKjorXiyrmi7qXv6dSvqRioajScknlm2VVblZUvVNV5U5Fvu/ruUvPjbvZAAAAALBpEXIAAAAAq3KHc3rmB8+odKuklQ9WVMw3wo5SodQIOW6UVb5ZVrFQlFNy9OyFZ5VZYjMOAAAAABgXlqsCAAAA1ll4bEHP157X+afO673vv6fETEK+11iLyjANuZarA08d0PHvHh9zSwEAAAAAhBwAAABAGydeO6ETr53Q8jvLyr+dl9SY6ZF5lJkbAAAAABAXhBwAAABAF5mDGWUOEmwAAAAAQBwRcgAAAAARMgxj7b3v+6Gu63Ru/fFe57vVDQAAAAAbBRuPAwAAABFphgzNgKFdKNHrOsMw5Pv+2qs1uFj/ar1np3IAAAAAsFExkwMAAACIUDN86BU0dLou6AyMZqgxiMLlgvKXV/cbOZRTdinb9rq9L4e7/7XTvcteO937Pq336FUmijqD6FRP0Pu3Kz+M/oi6vmHVOWh/drtfv8/MsJ+xUX0XUT+jwy7X7T5R/d4CAICNh5ADAAAAiJlhLzt17uQ5XTl/RcktScmQtFqFU3G0/8n9Ovn6ycjr7GTvy/0PesdBt7Y1z4UZfB5Gf/Rqa9TtDFPnoP3ZT/1hrotS1H3aqdwgfTqMZ22c9wIAABsbIQcAAAAQM637bHRamqq1TK8lqq5fuq4zR88ovSutxSOLSs2mZJiGPMeTXbJlrVi6euGq10TRwgAAIABJREFUXpp+SafeOqXc4Zyk3gOhQQafuw3C9hrcbZ7rd9Bz2H/53atfggw+rz8/6v4Ydjvb1dl6317X9ltfWKN6xsL2adhyg/bpoP1CUAEAAEaBPTkAAACADaLTfh1SI+B49euvKruU1b7H92nhsQXlDuWUeTSj3Z/drR0P7tD8vnntfGin0nvSOvvEWX34kw+H2t4gA+RxXaImyMBwv/cYRn90C6KG1c5e4Ve/bYnyM7e79ziesX77NEy5Qfo0bL9E1Z9hZu8AAIDNi5ADAAAAiFBzNkWvWRWdrotiw/B2Mz3OHD2jHQ/sUHYpq9yhnO7/yv3Kfimr3Z/bre0PbNf83nltXdiq2cys5rJzSt6X1CtHXhm4Lfi4oAHAqP8CvrW+uLYzqDgOjoft00n/LsKK43cIAADiieWqAAAAgIg0l4xqBhWdlp3qdl2vjcg7bTjercy5k+eU3p3Wzod2atcju5RZymh+77zqdl3lj8pKzCRkGIY811Pdrsu1XDkVR9Y9S2988w0d/97xsF0yVlFtfozgxv0X+OMOh3jGBrfRwhoAADB8hBwAAABAhDptFN5uD41+79HP/de7cv6KFo8sKr0nrbmFOc3vnde2xW2yVizV7bqciiO7aCs1m2q80ikl00nNbJ/Ru6+/O7SQY1wD0sMeiGaQ9teiCAH63UR8nEFDHNoQRFyf0UnpPwAAEC+EHAAAAMAGVrhcUHJLUqnZlJJbkjKTpup2XdaKJddy5Xurs0qmDJkJU2bSlJkylUglGq+ZhJbfWVbmYGagdnQbVB3GgGanTbZHKQ4DtddONz57uw2mh9knne49yCB22A3uhyUOz9ig4vCMtopjmwAAQLyxJwcAAAAQgYcffliFQmHt1bT+WK/jV65cWTt/8eLFSN7/6LUfSYZkmIZ8z5dTdlT+qKxivqjKrYrski3XcuW5nnzPl/zVpa9MyZhqlMm/nR+wd0ar18bWoxjcj+NAbTPsaL5GobkRddgNqYP256gDhnE+Y4OK6zMa5z4DAADxxkwOAAAAYEAvvPCC8vl82yWj+lleqnVvjijeNw5InuPJKTmq3qkqMZOQU3FkGIZcy1XtXk1O2ZFruao79UbgUfcboUdEOg2odpplMAzNWQ3DEtfB404bUw97w+pOQUDQ77zfgCMO/T7sZ2xQceqr9eLaLgAAMBkIOQAAAIAI5HK5gY/v379/7f2xY8eief/0Mb3/r96XXbJVXalq+ua0DMOQXbRlTDU2G3fKjmr3arLLtpzKx8MOwzSUO9z+M0Shn0HvOJuEQdo4t61VmP7sFi5MwvczbJPQB3yHAAAgDEIOAAAAYAPLLmXlVBxZK5YqtypKTCfkuZ5SsymZCVO+58u1XNllW7W7Ndml1aCj4sq1Gq/Mo4Ptx7HRTfLgaxzbHsc2TTr6FAAAbGSEHAAAAMAGt//J/bp64Woj2JhqbDyemk3JTJqSL9WdupyKI7vUCDqaszqsu5YOPHVg3M2PzDCWEep38LjXzJW4DEaHbeegyzWF+fzdrh11f3b77IP2adhnJi7PVDdx+g4BAMDkYeNxAAAAYIM7+fpJORVHxUJRpeWSivmiih8WVcw3fi7fKKtys6Lq7aqqd6qq3a3JumtJnnT8u8eH2raog4dO91t/PKrB0kEHX1vbOqy9HHr1Sb+begdtZ7vrutU5KYPZUTxjUfVpr3KT0qcAAACDYCYHAAAAsAmceuuUzj5xVsXrRTkVR8l0UolUQjIlv+6r7tTlVty1GRy+7+u5S89FVn+vwdhug969jreWHcXGz+vr6PXX++2ONcu0K9vrL/J7He+nL7sJ285e5doZpD8HMepnbFh92us777dPo+6XIM8oAABAWMzkAAAAADaB3OGcnvnBMyrdKmnlg5XGbI58UaVCaW02R/lmWcVCUU7J0bMXnlVmafh7cVw7He2AZ7d7RV3XILoNZo+yrl71hW3nIHXG3aDP2DD6FAAAYDNjJgcAAACwSSw8tqDna8/r/FPn9d7331NiJiHf8yVJhmnItVwdeOpA4CWqggyuDjIAG7bsKAZ9o6ij33uMoy8HKRvVvhqDCBscDHrfYZUfVZ/G6XePEAcAAPRCyAEAAABsMideO6ETr53Q8jvLyr+dl9SY6ZF5dPgzNwAAAAAgSoQcAAAAwCaVOZhR5iDBBgAAAIDJRcgBAAAAYNPrZyNpls8BAAAA4oONxwEAAAAAAAAAwERiJgcAAACATY/ZGQAAAMBkYiYHAAAAAAAAAACYSIQcAAAAAAAAAABgIhFyAAAAAAAAAACAiUTIAQAAAAAAAAAAJhIhBwAAAAAAAAAAmEiEHAAAAAAAAAAAYCIRcgAAAAAAAAAAgIlEyAEAAAAAAAAAACYSIQcAAAAAAAAAAJhIhBwAAAAAAAAAAGAiEXIAAAAAAAAAAICJRMgBAAAAAAAAAAAmEiEHAAAAAAAAAACYSIQcAAAAAAAAAABgIhFyAAAAAAAAAACAiUTIAQAAAAAAAAAAJhIhBwAAAAAAAAAAmEiEHAAAAAAAAAAAYCIlxt0AAAAAABubYRhr733fH+g6wzA+cS5sOQAAAACTj5kcAAAAAIamGUA0A4b1gUS/13U71m85AAAAABsDMzkAAAAADFUzgPB9v2vg0O265kyMduXDlmuncLmg/OW8JCl3KKfsUrbtdXtfDnS7T7h2unfZa6f7u+f6+3Uq26nOfupqd49+29rPvfu5f9i2tZYbdX3DrDOK77zdvXqV77dPB/19GNV3EfUzGraNg9TZT1kAACYJIQcAAACAWAu71FQ/5c6dPKcr568ouSUpGZJWizkVR/uf3K+Tr5/su/6w9r482CBmP9c0z4UdiO6nrf3ee/25MIPP3doWNqTq1dao2xm2zii+8yD3CnNdlKLu02H057CetW51disfxe8sAABxQ8gBAAAAILaGHXBcv3RdZ46eUXpXWotHFpWaTckwDXmOJ7tky1qxdPXCVb00/ZJOvXVKucM5Sb0HNIMMInYbTI16ILJXe4MGAuuvibKtYds3aNua5/odiG6957Db2a7O1vsGub7fOsOIqk97CdunYcsN2p9h+mVcvxcAAEwa9uQAAAAAEGuGYay9mj9HUe76pet69euvKruU1b7H92nhsQXlDuWUeTSj3Z/drR0P7tD8vnntfGin0nvSOvvEWX34kw+j/XAtwiwfFWZwPEx9rddGNUgaRfv6bdu10+EH1fsZAO91XdAZNJ2u6/d4kDr7qb/1vuMYOA/7XPZTbpD+DNsv4/i9AABgUhFyAAAAABiqoOFEu+t83//Yq3ksinJnjp7Rjgd2KLuUVe5QTvd/5X5lv5TV7s/t1vYHtmt+77y2LmzVbGZWc9k5Je9L6pUjr4TpgqEZ9qBl0EH2cS5VFLe2tdYX13YGFceB8bB9OunfRRCb4TMCANCK5aoAAAAADE1z0+9mALE+aFi/pFS368Lev5tzJ88pvTutnQ/t1K5HdimzlNH83nnV7brKH5WVmEnIMAx5rqe6XZdruXIqjqx7lt745hs6/r3jgftgGBig3Pj63TtjWPWPq744hisAACCeCDkAAAAADFWn4KH1eJCAot01YcpdOX9Fi0cWld6T1tzCnOb3zmvb4jZZK5bqdl1OxZFdtJWaTTVe6ZSS6aRmts/o3dffHVrI0c9G4lEMAsc9LIl7+0YpqhBg1M9YWHFoQxBx2nAdAIDNipADAAAAwKZSuFxQcktSqdmUkluSMpOm6nZd1ool13Lle6uzQ6YMmQlTZtKUmTKVSCUar5mElt9ZVuZgZqB2hNk0Ouj5fsV9IDkO7bt2uvGdtdu0eZiDzp3uPWgIMOpnrJ+6JnEQPw77kQAAsFmxJwcAAACAoSgUCh97hT3+i1/8Yu39xYsXB36fv5yXDMkwDfmeL6fsqPxRWcV8UZVbFdklW67lynM9+Z4v+av7fZiSMdUok387P2j3hBLl4G/c/1I+zu1rhh3N1yg0N7AeZIPvIH066oCh10bucQ48xvGMxvn3AgCAcWEmBwAAAIDIvfjii3rwwQd17NixT5wLunxVu+NRvZcveY4np+SoeqeqxExCTsWRYRhyLVe1ezU5ZUeu5aru1BuBR91vhB4R6TRI2Wu2QJTLVMV1oDSu7eu0afOwN3PuFAR0elba6SfgiEO/Nz9fXBFwAAAQH4QcAAAAAIbi5z//uZ5++ulPHM/lcm2v73T8wQcfXHu/PjQJ+z53qFGPXbJVXalq+ua0DMOQXbRlTDU2G3fKjmr3arLLtpzKx8MOwzSUO9y+rVHoNXjdbeB30gay24l7+6R4t62dfvt00GdsoyPgAAAgXgg5AAAAAGwq2aWsnIoja8VS5VZFiemEPNdTajYlM2HK93y5liu7bKt2tya7tBp0VFy5VuOVeXSw/TjGJe4DpXFvXzdxbXtc2zWpCDgAAIgfQg4AAAAAm87+J/fr6oWrjWBjqrHxeGo2JTNpSr5Ud+pyKo7sUiPoaM7qsO5aOvDUgbG0edDZGWEGSoPOKhnHElqjbNsgwrYziuWawvRpVPcaVLfPPmifhn1mJiHgmJTfCwAAosTG4wAAAAA2nZOvn5RTcVQsFFVaLqmYL6r4YVHFfOPn8o2yKjcrqt6uqnqnqtrdmqy7luRJx797fKhtG8Y+BFEMbLa2K06boA+zbUHuG7T9YdvZ7ro4Dsj3q1d/SqPr017lJiHg6FS+088AAGwUzOQAAAAAsCmdeuuUzj5xVsXrRTkVR8l0UolUQjIlv+6r7tTlVty1GRy+7+u5S89FVn+vAceoBlPX19Prr+M7HW+Wa1d+0HYO0r6wbQsyuN6pfJiB4rDt7FWuk0G/8zDC9mnYgfdh9Wmv77zf/oyiX0b1ewEAwKRiJgcAAACATSl3OKdnfvCMSrdKWvlgpTGbI19UqVBam81RvllWsVCUU3L07IVnlVka/l4c107HbxCy24DxuI2ybd3q6lVf2HYOUmfcdWv/uPp0o9gMnxEAgCZmcgAAAADYtBYeW9Dzted1/qnzeu/77ykxk5Dv+ZIkwzTkWq4OPHUg8BJVQQYQRzn4HmV9wxocjeK+/d5jkDrHUXaUny/sPUf12cZR7yR858O6BwAAk4CQAwAAAMCmd+K1Ezrx2gktv7Os/Nt5SY2ZHplHhz9zAwAAAEB4hBwAAAAAsCpzMKPMQYINAAAAYFIQcgAAAAAAItPPRtIspwMAAIBBsfE4AAAAAAAAAACYSMzkAAAAAABEhtkZAAAAGCVmcgAAAAAAAAAAgIlEyAEAAAAAAAAAACYSIQcAAAAAAAAAAJhIhBwAAAAAAAAAAGAiEXIAAAAAAAAAAICJRMgBAAAAAAAAAAAmEiEHAAAAAAAAAACYSIlxNwAAAADYLAzDWHvv+36o68KcW3+8U91B2wYAAAAAccJMDgAAAGAEmiFCM0BoDR6CXNfrnO/7a692wUbz1a7ObucBAAAAIK4IOQAAAIARaQYIvYKEbtcFvUdQzYADAAAAACYRy1UBAAAAG0Dr7I0olrlq9aurv9Jf/l9/qffffF93rt6RJO14YIf2/f19eux/fEzzn54f6DMAAAAAQL8IOQAAAIANoHVGxvqf24Ua6491KrfehX9yQT/9v38q+dLM/Iy2f3q7vLqnyo2KLv/ry3rnlXd06PcP6Wt//LWoPxoAAAAAdETIAQAAAKCr7zz+HRV+WlDm0Yxm98wqlU5JkhzLUe1eTdYdS5VbFf2n//M/6cY7N/T0nz095hYDAAAA2CwIOQAAAIARac6S6LTpeJDrgt4jKmefOKvlny1r77G92vngTs1mZ5WYSahu11W7W1PldkWVuYqmZqZkJAxde+uaXv1vXpUOj6R5AAAAADY5Qg4AAABgBJrBRDOc6La0VKfrgpxb//P6+7e2JUi5/3z+P+v6X1zX4lcX9anPf0p7DuzR1vu3SoZkrVgqf1RWYiYhwzTke748x1PdruuDH30gbZX0UN/dBAAAAAB9IeQAAAAARqTTpt6tx7tt/h3mXLcy3c6/+fyb2r5vu7YtbtOO39ih3Z/frZ0P7VT1dlXFfFG+56tu1+VUHTkVR3bZll2yVUvX5PzQIeQAAAAAMHTmuBsAAAAAIH5KhZLu/Jc72vKpLbpv+32anp9e24vDTJiamp5SYibxydd9CSXTSemOZJRHs6QWAAAAgM2LmRwAAAAAPiF/Ob8WZhhThjzXk7ViSYbkuZ5cy5XnepLfWA7LMA2ZU6bMRONlmIb8fPcZJAAAAAAwKGZyAAAAAEN08eJFXb58ee31s5/9TC+++OLa+bi+X/kvKzKnzMaSVLXGJuOl5ZKK+aIqH1VUW6nJKTtyLVd1py6/7sv3/LXQY8qYku4E6aHJ1twjJchm8r2u67TRfLdyQesHAAAANipmcgAAAABD9KMf/Ui+7+uP/uiP1o594QtfWHv/wgsvxPL9/GfmGzM2qq5q92qq3KrInDJll2yZU6bqdl12qbEHh1P5ddjhuZ4811Pdq0s7uvXM5Fu/AXwzaGi3v0mQ6zoFHJ02qG/3MwAAALAZEXIAAAAA+ITcl3Ly6p5q92qq3qkquSUp3/OVKqZkJkz5dV9O1ZFdtBuvsi234jbCDrsu3/Pl5zb+AHwzZGgGGGGua4YV/czGCBNwFC4XlL+clyTlDuWUXcq2vW7vy33dds21073LXjvd+z7t7tGtXBR1BtGpnqD37/dzdSo36vqGVeeg/dntfr3u0W+fDvqMjeq7iPoZHfaz1u0+Uf3eAgA2B0IOAAAAAJ8wm53Vzt/YqeqtqsrbyjITpupOXanZlKaSU41lrOy67LIt+97qa3VWh1N1pF2S0uP+FPHXLaxoDT66zf7oFnicO3lOV85fUXJLUjIkrV7qVBztf3K/Tr5+MvwH6NPel/sf9O5VbhR6tU0KN/gctj+6GaQfw7QzTJ2D9mc/9Ye5LkpR92mncoP06TietajaAACARMgBAAAAoIPf+ue/pfNPn1dyLinDMOTW3LWQQ5I8x5NjObJLtmr3arLuWbJLttyqK/32mBs/AXrNxui1PFWvpauuX7quM0fPKL0rrcUji0rNpmSYhjzHk12yZa1Yunrhql6afkmn3jql3OGcpN4DoUEGLrsNwvYa/Ow0IN5vuaj16pcgg8/rz/f7ufodBO63HwdtZ7s6W+/b69p+6wsrqj7tJWyfhi03aJ+O6lkDACBqbDwOAAAAoK2H/8HD+vTRT+vutbsqFooq5osqfrj6b6Go0o2SKjcrqt6uyvqVpdpKTZXbFX3m8c9ID4679ZOhdePwqDYQv37pul79+qvKLmW17/F9WnhsQblDOWUezWj3Z3drx4M7NL9vXjsf2qn0nrTOPnFWH/7kw0jq7iTIX633M3A7SlGEK63XBQkMwg6qD9KP/bazV/jVb1ui/Mzt7j2O56nfPg1TbpA+HdezFuReAAAEQcgBAAAAoKOn/9+ntfj3FnX3b++qlC81Ao58UaVCSaUbJZVvlFW+WVblVkXlW2X9xtd+Q9/8f7457maPTNBwot11vu9/7NU8FoUzR89oxwM7lF3KKncop/u/cr+yX8pq9+d2a/sD2zW/d15bF7ZqNjOrueyckvcl9cqRVyKpe5gmbUmboAHAqD9Xa31xbWdQcRwcD9unk/5dtArazjh+hwCAycFyVQAAAAC6eubPntEP/+cf6u1/+bZKN0qamp5SYrrxvxKu5apeq2vqvin93X/yd3XsxWPjbewINffMaAYXnZaP6nZdkPuv/znIuXMnzym9O62dD+3Urkd2KbOU0fzeedXtusoflZWYScgwDHmup7pdl2u5ciqOrHuW3vjmGzr+veMhemP8wm6ajPDG/Rf44w6HeMYGNylhDQAg3gg5AAAAAPT0tf/lazr0e4f0l//yL/X+D9/X7b+5LUna8dAOfeZrn9Fj/8Nj2nr/1jG3cvS6bRoe5Lpe13Qr1+nclfNXtHhkUek9ac0tzGl+77y2LW6TtWKpbtflVBzZRVup2VTjlU4pmU5qZvuM3n393aGFHOMakB72QDSDtL8WRQjQ7ybicdiQPu5hR1yf0UnpPwBA/BFyAAAAAAhk295tevx/fXzczUAXhcsFJbcklZpNKbklKTNpqm7XZa1Yci1Xvrc6q2TKkJkwZSZNmSlTiVSi8ZpJaPmdZWUOZgZqR7dB1U5r+O99uf1GxUEGaMOUiVocBmoH7cewOt17kEHssBvcD0scnrFBRdlfUT1rcfi9AQBMPkIOAAAAAOhToVD42M/ZbDaS48vLyzp48KAk6eLFizp69Ghf72f/elYyJMM05Hu+nLKj8kdl1e26fM+XXbLlWq4815Pv+ZK/uk+IKRlTjTL5t/MDhxyD6HfwuFto0rzfsAZS4/yX6HEIevptQ9D+HPVnG+czNqhRPKNhvo9JDIkAAPHFxuMAAAAA0IcXX3xRb7755ic2DZfabybez/HWc6He+5LneHJKjqp3qiotNzaML98oq3q7qtq9mpyy09hPxak3Ao+63wg9InLtdPuX1Hlwc/01QY4HacMwxTXgiLof+6m307EgA9r9Bhxx6Pc4tKGbYfdV2GctTt8hAGBjYCYHAAAAAPTp5z//uZ5++ulPHM/lcm2vD3p8/c/Hjh3r+31ha2NmiF2yVV2pavrmtAzDkF20ZUw1Nht3yo5q92qyy7acysfDDsM0lDvcvq1R6LbEzfpr4m4SBmnj3LZWYfqzW3AyCd/PsI2yD8LWwXcIAIgKIQcAAAAAbBDZpayciiNrxVLlVkWJ6YQ811NqNiUzYcr3fLmWK7tsq3a3Jru0GnRUXLlW45V5dHxLVXUSpwHPOLWlX3FsexzbNOni0KdxaAMAYPMg5AAAAACADWT/k/t19cLVRrAx1dh4PDWbkpk0JV+qO3U5FUd2qRF0NGd1WHctHXjqwLibH5lhrPnf78Btr5krcRkIDtvO9ftShBHm88dpGaRun33QPg37zMTlmeomTt8hAGBjYE8OAAAAANhATr5+Uk7FUbFQXNuPo/hhUcV84+fyjbIqNyuq3q6qeqeq2t2arLuW5EnHv3t8qG0LsjxNp+PdBny7HY9qsHTQwdfWtg5r4+Uw/ditfNB2trsuyHcX98HsKJ6xqPq0V7m4hDyT8t0CADYOZnIAAAAAwAZz6q1TOvvEWRWvF+VUHCXTSSVSCcmU/LqvulOXW3HXZnD4vq/nLj0XWf29BmN7/RV6lHVFYX0dvf56v92xZpl2Zfvti9bj/YQ/3YRtZ69y7QzSn4MI26dhn7Fh9Wmv77zfPh31swYAQNSYyQEAAAAAG0zucE7P/OAZlW6VtPLBSmM2R76oUqG0NpujfLOsYqEop+To2QvPKrM0/L04rp3uf8PxMGV6lRu1UW6yHqYfg5QdVp1xN+gzNow+jYuN/L0DACYLMzkAAAAAYANaeGxBz9ee1/mnzuu977+nxExCvudLkgzTkGu5OvDUgcBLVAUZtBx0YDNM+VEMpkZRR7/3GKTOcZSNal+NQYQNDga977DKj6pPx1V2FPcDAGwOhBwAAAAAsIGdeO2ETrx2QsvvLCv/dl5SY6ZH5tHhz9wAAAAAho2QAwAAAAA2gczBjDIHCTYAAACwsRByAAAAABgqwzDW3vu+3/d13cr3unfQurG59LNZMsvnAAAAxBsbjwMAAAAYmmbI0AwY1ocOQa/zfb9jQNHtnGEYa+d93+9YNwAAAIDJxUwOAAAAAEPVDCF6BQ1Br+u33n4ULheUv7y6b8WhnLJL2YHbgfhhdgYAAMDGQcgBAAAAYMMKulzVuZPndOX8FSW3JCVD0uqlTsXR/if36+TrJ4fcUgAAAABhEHIAAAAA2LBa9/ZoDTquX7quM0fPKL0rrcUji0rNpmSYhjzHk12yZa1Yunrhql6afkmn3jql3OHcqD8CAAAAgC4IOQAAAABsStcvXderX39V2aWsdj60U+k9aSW3JOV7vpySo+pKVZVbFaVmUyoWijr7xFk984Nnxt1sAAAAAOsQcgAAAAAYquYMil77bAS9rt96Ozlz9IyyS1lll7La9cguzS3MyUyacsqOqneqmr45rcR0QuaUKUkqXi/qlSOvSH8QSfMAAAAARICQAwAAAMDQNEOLZnDRafmoXte1vm+e73auNTBZf89zJ88pvTutnQ/t1K5HdimzlNH83nnV7brKH5WVmEnIMAx5rqe6XZdruXIqjqx7luxztsQWHQAAAEAsEHIAAAAAGKpOsylajwe9Lui5buevnL+ixSOLSu9Ja25hTvN757VtcZusFUt1uy6n4sgu2krNphqvdErJdFIz22dkX7G71gkAAABgdMxxNwAAAAAARqlwuaDklqRSsykltyRlJk3V7bqsFUuu5cr3VmeVTBkyE6bMpCkzZSqRSiiRSkgJyViOZkktAAAAAIMh5AAAAAAwFA8//LAKhcLaS5J+8pOfrJ2/ePHiWN7/6LUfSYZkmEZjk/Gyo/JHZRXzRVVuVWSXbLmWK8/15Hu+5K8uhWWqEXwYppTvuzsAAAAADAHLVQEAAACI3AsvvKB8Pt91SapxvW8ckDzHk1NqbDKemEnIqTgyDEOu5ap2ryan7Mi1XNWdeiPwqPuN0AMAAABAbBByAAAAABiKXC73iWNf/vKX194fO3ZsPO+fPqb3/9X7sku2qitVTd+clmEYsou2jKnGZuNO2VHtXk122ZZTaQk7fF9+jrADAAAAiANCDgAAAACbSnYpK6fiyFqxVLlVUWI6Ic/1lJpNyUyY8j1fruXKLtuq3a3JLq0GHRW3sWeH60uZcX8KAAAAABIhBwAAAIBNaP+T+3X1wtVGsDHV2Hg8NZuSmTQlX6o7dTkVR3apEXQ0Z3VYdy3p8+NuPQAAAIAmQg4AAAAAm87J10/qpemXVCwUJUmu5SqVTslMmTKMxpJVruXKqTSWrardrTUCDk/SCUmsVgUAAADEgjnuBgAAAGDWgdSpAAAgAElEQVTzMQxj7RX2ujDn1h9vPd/tHDamU2+dklNyVLxeVDFf1L38PZXyJRULRZWWSyrfLKtys6LqnaoqdyryfV/PXXpu3M0GAAAAsA4hBwAAAEaqGR74vv+xn/u5rtc53/fXXuvPrT/eLBvkHDam3OGcnvnBMyrdKmnlgxUV842wo1QoNUKOG2WVb5ZVLBTllBw9e+FZZZbYjAMAAACIE5arAgAAwMg1Q4TWEKKf64Leo5NmGNLvuUal0gcXP9D7P3xf1398XZJ0/2/er31/f58+ffTTEpNAJsbCYwt6vva8zj91Xu99/z0lZhLyvdXwzDTkWq4OPHVAx797fMwtBQAAANAOIQcAAADQh1/++S/1b5/6t7JWLBmmodSWlHzP1/UfX9eP/+THmtkxo2+8/g3lvpwbd1PRhxOvndCJ105o+Z1l5d/OS2rM9Mg8yswNAAAAIM4IOQAAALChBJnZEXYWx5//b3+u//BP/4PmPzOvPQf2KDWbkmEYcmuu7KKt6kpV5RtlnfmtM3r8jx/Xl09/eeDPg9HKHMwoc5BgAwAAAJgUhBwAAADYcNaHFFFtIH7xn17Uj//kx7r/N+/Xjgd2aDYzq+R9SXmup1qxpuqdqqZvTSt5X1KJ+xJ68w/flFNxIqkb8dW650uY63rdo9P5oHUDAAAAGxkhBwAAAEauOVsi6IyLdtcFuUfPvTUCWnl/Rf/xn/9HLf7movZ8YY92f263ti5slTFlyC7aqtyuKJVOaSo5JUnyPV+e6+lHL/5I+u8lzQ3cBMRQ89lrPoednrdu17WW6fVzv9cBAAAAGx0hBwAAAEZq/UBv8+em9QO13a7rdY/117UKs1TVv/udf6f5vzOv+X3z2vnQTmW+mNH2z2yXXbJVulGSkTDk133V7bpcy5VbdeWUHdkrtipvVKRnA3cPJkzr89rvdb1mf3Q6HybQKFwuKH95db+RQzlll7Jtr9v7ct+3liRdO9277LXTve/T7h7dynWqM0hd/Ri0nn4/V6dyo65vWHVG/b2tv1+ve/Tbp4M+16P6LqJ+Rof5rI3q9xYAsDkQcgAAAGDkgg7cdhvIDTv42+897ZKt6z++rsUji9qya4u27N6i9KfSmtk+IxlSspRUaktKyS3JX7/uSyqxJaHk1qT0t5LhRrNkFjafKJakOnfynK6cv6LklqRkSFq9jVNxtP/J/Tr5+skIWhrM3pf7H/TuVq5XGSmaQdNB6+lUPmx/dBOmH3uVDVuuU9mov7egfRW2TwcRdZ8O43dh1M/aqH5vAQCbByEHAAAA0EXhckFT01ON4GI6IUOGaqWayjfKqtt1eY7XGHw2JMM0ZEwZMpOmppJTmkpOyZgy5H3ojftjYAK0m7kRZumqpuuXruvM0TNK70pr8ciiUrMpGaYhz/Fkl2xZK5auXriql6Zf0qm3Til3OCep98BkkMHHboOwvQZ3Ow2Idys3SH1B9eqXIIPP68/32x/9DkT324+DtrNdna337XVtv/WFFVWf9hK2T8OWG7RPR/WstSvTT1kAAFqZ424AAAAANodr167pZz/7mX7+85+vvX7605+unf/TP/3TWL6/8bMbMk1TMqS6U1et2Ag4ivmiyrfKqt2tyak4jcCj7sn3/I+FHqZpyrjBTA50F3ZPjW4Bx6tff1XZpaz2Pb5PC48tKHcop8yjGe3+7G7teHDH2vJr6T1pnX3irD78yYdRfJSOgvzVej8Dt81z/Q6ihtFPKBP0uiCBQdhB9X77sdt1QZcd6ud7iKI/g9bfeu9xDJz326dhyg3Sp6N+1qL8/gEAkAg5AAAAMCLf/va3dfHiRWWz2bXXvn371s4fPXo0lu+37Noir+7JtVzZRVvV21WVlkuNkGO5rMrtimp3a7JLttyq++vZHa4vv94IPPwtbAi9UTWXk+q2H0ev66IOOCTpzNEz2vHADmWXssodyun+r9yv7Jey2v253dr+wHbN753X1oWtms3Mai47p+R9Sb1y5JW+2zBq41huaBBBA4BRf67W+uLazqDiODAetk8n/btoNSntBABMNparAgAAwMjcvn1bc3Nzbc99+tOfjuX73KGcfN9vBBx3qkrNpho/z9mNpahcT27VVe1eTbViTU7ZaWw+XnNVd+ryPE/GAjM5NqLmJuLN4KLT0lK9rlv/7/rz3TYp71bu3MlzSu9Oa+dDO7XrkV3KLGU0v3dedbuu8kdlJWYSMozGs1u363ItV07FkXXP0hvffEPHv3c8gt7BZjDu/RPGHQ7FMVwBAGAzIuQAAAAAupjfN6/UbErWHUuVWxVNpabkuZ5qxZqmElPyPV9uzZVTdtaCDrtsy6k6cqqONC1pftyfAsPSaSZFt701ghwfpNyV81e0eGRR6T1pzS3MaX7vvLYtbpO1Yqlu1+VUHNlFW6nZVOOVTimZTmpm+4zeff3doYUc4xqQHvZANH+p/mtRhAD9biI+zqAhDm0IYpKe0UlqKwAgPgg5AAAAgC7MhKnDv39Yf/Enf6HkbFKGaahu15VMJzWVmpL8xl4dbtWVXbJVu1tT7V5j+Sqn5Ej/leQbLFeF0ShcLii5JanUbErJLUmZSVN1uy5rxZJrufK91VklU4bMhCkzacpMmUqkEo3XTELL7ywrczAzUDu6DVR2WsN/78vtNxvud9C7V13DEoeB7ij6MYxO9x4kBAi7wf2wjLI/hyXqfWqG+azF4fcJADA52JMDAAAA6OHYi8e0ZdcW3bt+T6VCYz+O5qu0XFL5Rlnlm439Oaq/qqq2UlP1TlVzC3PSV8fdegzDww8/rEKhsPZqWn9s/fGLFy+O5H3+cn5t03vf8+WUHZU/KquYL6pyq9LYO8Zy5bmefM+X/NUlr0zJmGqUyb+dD90vUWgOnDZfg95rmOL8l/xR9mNQzQ2sw27wHbQ/Rx0w9NpcO86Bxyie0aietTj/PgEA4o2ZHAAAAEAAv//e7+tfPPQvtPLBitLV9NpMjuZgct1uzOZwyo4qdyqamZ/RP/qrf6Q//t//eNxNR8ReeOEF5fP5tktGBVleatjv5Uue48kpOareqSoxk5BTcWQYhlyrsX9Mc++YulNvBB51vxF6RKTTIGWnv/xeX6bTkke9Bk/b3bNbfYOK64DsoP04aL2tx4J+B/0GHHHo9+bni6th91WUz1qcvlcAwOQh5AAAAAACSKVT+sd/+4/1b778b3Tnb+7IMI1G0JGYku/78mxPdsWWfOlT+z+lb/34WxL7jW9YuVyur+PHjh0byfvcoUb9dslWdaWq6ZvTMgxDdtGWMdXYbLy5f4xdtuVUPh52GKah3OH2nyEKQQa9R7WkziAmYUA2zm1rFaY/uw2gT8L3M2yj7INB6+D7AgAMipADAAAACMiYMvR7b/+e/urbf6VL/8cl3f75bZnJxgqwnuNp58M79Zv/02/qi7/zxTG3FJtVdikrp+LIWrFUuVVRYjohz/WUmk3JTJjyPV+u5couN/aPsUurQUfFlWs1XplHB9uPYxjiNAgap7b0K45tj2ObJl0c+nQSZ+YAACYXIQcAAADQpy/+7hf1xd9tBBl3/uaOJGnHgzvG2SRgzf4n9+vqhauNYGOqsfF4ajbVCOR8qe7U5VQc2aVG0NGc1WHdtXTgqQPjbn6s9Tsg22smSVwGeMO2c9DlmsJ8/m7Xjro/u332Qfs07DMTl2cqiElqKwAg3th4HAAAABjAjgd3EHAgVk6+flJOxVGxUFRpuaRivqjih0UV842fyzfKqtysqHq7quqdqmp3a7LuWpInHf/u8aG2LcgSQ52OdxvwDVNfvwYdkG1ty7D2cgjTj93KB21nu+uCfHdxH+Du1Z/S6Po06PM+7pAnSDsm5fsHAEwGZnIAAAAAwAZz6q1TOvvEWRWvF+VUHCXTSSVSCcmU/LqvulOXW3HXZnD4vq/nLj0XWf1hNgkPUi5smaj2DOhVX7fNtzuV7bcvOm3yHKRsN2Hb2atcO4P05yDC9mnYQGpYfdrrO++3T0f9rI3r+wcAbFzM5AAAABPNMIy1V5jrepUPUi5I/QAwSrnDOT3zg2dUulXSygcrjdkc+aJKhdLabI7yzbKKhaKckqNnLzyrzNLw9+K4drr/DcfDlAlSdpRGscl6kLqC9Fc/x6OoM+66tX9cfRoXG/l7BwBMFmZyAACAidUMFnzfXwsafN/v67rmv+1Citb7tSvXWgcAxMXCYwt6vva8zj91Xu99/z0lZhLyvdX/5pmGXMvVgacOBF6iKsig5aADm2HKj2IwNYo6+r3HIHWOo2xU+2oMImxwMOh9h1V+VH066rIEIACAqBFyAACAibY+dOgWNAS9rl2ZXjqFKx+7V91X/u28Pnz7Q0nSwuEF5b6UkzFFOAJguE68dkInXjuh5XeWlX87L6kx0yPz6PBnbgAAAADDRsgBAADQxfpAJGjo0ep7/9339Nf//q+VvC8pQ4Z8Ne7jVB098tuP6BvnvxFJWwGgm8zBjDIHCTYAAACwsRByAAAAdNFpuapux5p++dYvdebYGc1+alaLf29R07PTMqYM1e267FJjs98P/r8P9M9S/0zPvfWcco/lhvpZAAAN/WyWzNI6AAAA8UbIAQAAMAS//PNf6tX/9lXlDuW088Gdms3MKrklKa/uySk7qv6qqurtqlLplErLJX3nv/6O/uGf/cNxNxsAAAAAgIlCyAEAACZacyZFr302gl7XrkwY3z72bWW/lFX2YFa7PrtLc7k5TSWnZFdsVW9XNX1zWomZhAxzdVN019crX31F+oNQ1QEA+sDsDAAAgI2DkAMAAEysZmjRDC46LS3V67rW9502KQ+6VNW5E+eU3p3Wzgd3atdndyl7MKttn96meq2u8s2yEtMJGYYhz/VUr9XlWq6ciiPrniX7nC2dHKhbAAAAAADYNMxxNwAAAGAQvu+vvVqPB72u9RWkXLs6mq68cUXbH9iu2cys5nJz2rZ3m7b9nW3asmuLZrbNaHrrtFKzqY+/0inNbJ+RroTpBQAAAAAANidmcgAAAESocLmg5JakUrMpJe5LyEyaqtfqsn5lya258r3VWSVThsyEKTPZeE2lppRIJaSEZCwHX1ILAAAAAIDNjJkcAABg4pimqc9+9rPK5/Nrr6Z33nln7f2bb7458vf5y/lGiGEa8j1fTtlR+WZZxUJRlVsV2UVbruXKcz35ni/5q8tkmWoEH4YpPx9uHxAAAAAAADYbZnIAAICJ8/zzz6tQKLQ9NzU1Ndb3zX066nZddslW9U5ViZmEnLIjwzTkWq5q92pyyo5cy1XdqTcCj7rfmOUhZnEAAAAAABAUIQcAAJhI2Wy27fEvfOELa++/+tWvjvx99kuNdjllR9aKpfJHZUmSPWfLmGpsNu6UHdXu1WSXbDmV1bDDboQdnu9JuV6fHgAAAAAASIQcAAAAkcouZeVUGgFH5VZFiemE/Lovu2jLTJjyPV+u5cou2Y2go7gadFRduZYr3/WlzLg/BQAAAAAAk4GQAwAAIGKf/8bn9Ysf/EKp2ZTMKVN1u954nzQlX6o7dTkVR3axEXTU7tVkl23V7takA+NuPQAAAAAAk4OQAwAAIGInvndCL02/pGKhKPmSa7lKpVMyU6YMo7FklWu5ciqNZatqd2uyViz5vi8dl8S+4wAAAAAABGKOuwEAAAAb0bf+4ltyyo6KHxZVzBd1L39PxXxRxUJRpeWSyh+VVblZUfVOVZXbFfmGr2/9+FvjbjYAAAAAABOFkAMAAGAIMksZ/c6F31H5dlkrH6yomC+qlC+pVCg1Qo4bZZVvllVaLsmpOvrdH/6u9nxxz7ibDQAAAADARGG5KgAAgCHJLGX0h9Yf6o2n39C733tXiZnE2lJUhmHIrbk68PQB/fZ3fnu8DQUAAAAAYEIRcgAAAAzZ8VeP6/irx3Xjr24ofzkvScodymnPF5i5AQAAAADAIAg5AAAARmTPF/ewJBUAAAAAABFiTw4AAAAAAAAAADCRCDkAAAAAAAAAAMBEIuQAAAAAAAAAAAATiZADAAAAAAAAAABMJDYeBwAAAEbEMIy1977vh76u0/lu5YLWDQAAAACThJkcAAAAwAg0Q4ZmwLA+dOjnOsMw5Pv+2itIudYyneoGAAAAgEnDTA4AAABgRJoBRK+godN1zbBi0PsHUbhcUP5yXpKUO5RTdinb9rq9L4e7/7XTvcteO93fPdffr1fZ1rr7vb6TftsctJ6g921XPkjZfvsj6vqGWeegfdrpXqN+xsKUH8Z3EfUz2s/30E/ZUf3OAgCA8SPkAAAAACZImGWnWkOPbuXOnTynK+evKLklKRmSVi91Ko72P7lfJ18/GardYex9ebCB70GuG4dubWueCzOo260fw/ZHr7ZG3c6wdQ7ap0HvFea6KEXdp8Poz0H6Jc6/twAAYPwIOQAAAIAJ0rpEVZCgo/W6duWuX7quM0fPKL0rrcUji0rNpmSYhjzHk12yZa1Yunrhql6afkmn3jql3OGcpN4DmkEGkLsNpvYTdPSjec8o/tq738HyMPX06o9OMwyC9mO//dGurlG0s12drfcNcn2/dYYRVZ/2ErZPw5YbtD/D9ku/ZUf1OwsAAMaPPTkAAACATe76pet69euvKruU1b7H92nhsQXlDuWUeTSj3Z/drR0P7tD8vnntfGin0nvSOvvEWX34kw+H2qYwS9gEDVTiOrAZZGC433sECQzCDqr3MwDe67qgSzF1uq7f40Hq7Kf+1vuO4xnrt0/DlBukPwfplzj/3gIAgPEj5AAAAABGpLlkVK/9MoJeF1W5M0fPaMcDO5Rdyip3KKf7v3K/sl/Kavfndmv7A9s1v3deWxe2ajYzq7nsnJL3JfXKkVf6qmPYxjUAGodldIIGAKNua2t9cW1nUHEcZA/bp5P+XQxiI34mAAA2O5arAgAAAEaguS9GM4DotHxUt+u67a0Rtty5k+eU3p3Wzod2atcju5RZymh+77zqdl3lj8pKzCRkGIY811Pdrsu1XDkVR9Y9S2988w0d/97xSPonrLgMWMZxAHyjGPfSQuMOh3i2hoN+BQBg4yDkAAAAAEak0/4Zrce77bMR9bkr569o8cii0nvSmluY0/zeeW1b3CZrxVLdrsupOLKLtlKzqcYrnVIyndTM9hm9+/q7Qws5ggwsj3vwe5TiEubEQVQhwKQ8Y3FoQxA8owAAYFwIOQAAAIBNqnC5oOSWpFKzKSW3JGUmTdXtuqwVS67lyvdWZ4dMGTITpsykKTNlKpFKNF4zCS2/s6zMwcxA7QizaXTQ88M0jsHnOAx0Xzvd+OztNpge5kB3p3sP+j3E6RkbZX8OSxye0U4mJTACAAD9IeQAAAAAhuzhhx9WoVBY+zmbzUrSx45FdTyRSOjKlSs6evSoJOnixYsd38/+9axkSIZpyPd8OWVH5Y/Kqtt1+Z4vu2TLtVx5riff8yV/db8PUzKmGmXyb+cHDjnCmMTB37DiPDA7ju8hiiAgSJ+O+rN12si92Y52oVJcxPkZBQAAGx8hBwAAADBEL7zwgvL5fNvlooIuX9Xv8db9OLq+9yXP8eSUHFXvVJWYScipODIMQ67lqnavJqfsyLVc1Z16I/Co+43QIyKdBkZ7zRbYDAOqcf2snTamHvaG1d2CgKAhQD8BRxz6fX3QEUdx6isAALA5EXIAAAAAQ5bL5UZ6/NixY4HeF7Y2ZoDYJVvVlaqmb07LMAzZRVvGVGOzcafsqHavJrtsy6l8POwwTEO5w+3bFIVeg9fdBn6HPfA6ioHdSRg8jnPb2um3T8f5jE2CSeqDSWorAADoDyEHAAAAsElll7JyKo6sFUuVWxUlphPyXE+p2ZTMhCnf8+VaruyyrdrdmuzSatBRceVajVfm0dEvVbUZTPKAbFzbHtd2TSr6EwAAxAUhBwAAALCJ7X9yv65euNoINqYaG4+nZlMyk6bkS3WnLqfiyC41go7mrA7rrqUDTx0YS5vHvczQsJcO6vczBJ3xMu7B6LDtjGK5pjB9GtW9BtXtsw/ap2Gfmbg8U0HFebkvAAAwOHPcDQAAAAAwPidfPymn4qhYKKq0XFIxX1Txw6KK+cbP5RtlVW5WVL1dVfVOVbW7NVl3LcmTjn/3+FDbFveByWEM8A46eNzaZ8Pqw073Ddr+sO1sd91GGJDv1Z/S6Pq0V7lJ6M9OJrHNAACgN2ZyAAAAAJvcqbdO6ewTZ1W8XpRTcZRMJ5VIJSRT8uu+6k5dbsVdm8Hh+76eu/RcZPX3GlSNcmAyyGBy1HUGtb4Nvf56v92xZpl2ZXsFAL2O97snSidh29mrXCeD9GlYYfs0bCA1rD7t9Z3325/DeNbi8HsLAADGj5kcAAAAwCaXO5zTMz94RqVbJa18sNKYzZEvqlQorc3mKN8sq1goyik5evbCs8osDX8vjmun4zdoGee/Yu82mD3KunrVF7adg9QZd93aP64+3Sji/DsLAACiwUwOAAAAAFp4bEHP157X+afO673vv6fETEK+50uSDNOQa7k68NSBwEtUBRlQHOXge1R1DnOgNIp793uPQeocR9lRfr6w9xzVZxtHvZP2vAxaFgAATAZCDgAAAABrTrx2QideO6Hld5aVfzsvqTHTI/Po8GduAAAAAEC/CDkAAAAAfELmYEaZgwQbAAAAAOKNkAMAAAAAhqCfjaRZUgcAAAAIh43HAQAAAAAAAADARGImBwAAAAAMAbMzAAAAgOFjJgcAAAAAAAAAAJhIhBwAAAAAAAAAAGAiEXIAAAAAAAAAAICJRMgBAAAAAAAAAAAmEiEHAAAAAAAAAACYSIQcAAAAAAAAAABgIhFyAAAAAAAAAACAiUTIAQAAAAAAAAAAJhIhBwAAAAAAAAAAmEiEHAAAAAAAAAAAYCIRcgAAAAAAAAAAgIlEyAEAAAAAAAAAACYSIQcAAAAAAAAAAJhIhBwAAAAAAAAAAGAiEXIAAAAAAAAAAICJRMgBAAAAAAAAAAAmEiEHAAAAAAAAAACYSIQcAAAAAAAAAABgIiXG3QAAAAAAk8EwjLX3vu+Hui7MufXHe9UNAAAAYHNhJgcAAACAnppBQzNgaA0eglxnGIZ83197tZ7rdv/15QAAAACgiZkcAAAAAAJpBgytAUXQ63oFFEHvH0ThckH5y3lJUu7/Z+/+g9w47zvPf7oHwAyJGXLIISUMRp4R9YOSTfNC0aTirE2TjpQouY33yiSVWNKulaU2d7V1uejuanNVexudzD15U7l/UtrbeJNzIq43tGSTRVWucskqxzg2d3XK2RajUqQwt9qlZdokhuLPGeJXo7uBvj96MIJG+NFoNH7NvF9VKGHQ/fTz4IsGXf5+8TzPnrSmd0/XPW/u+XDXv/B067YXnm7vmrXXa9W2Xt/t9tfu9dvpI+z4VrbrdX/d6rPTeDa7Xrv3Sph7q9P23fgsor5Hw8alnc+w299bAADWKoocAAAAAAZe0GLJycMnde7UOcXXxyVD0tKpTsHRjkd36PCJw10e6fvmnu8sYd7uue30F3Ys1WNhks/Nxhe22NRqrFGPM0yfncaznf7DnBelqGPaqF0nMQ0Tl259J6rHKHQAANAZihwAAAAAeq66dFUQ9fbuWPnaxdcu6tj+Y0puSWp236wS4wkZpqGKU5Gds2UtWDp/+ryeG31OR149ovTetKTWidAgycdmSdioE5iNfsEfdX+t4hIk+RxmfNVj7SaiGxUWujXOen2uvG6rc9vtL6yoYtpK2JiGbddpTMPEpdPverv3KQAACIY9OQAAAAD0VDsFjiAuvnZRL37uRU3vnta2h7dp5sEZpfekldqV0taPbtXmezdrctukprZPKXl7UscfOa5L37sUWf/1hPllfjttVp4bZYI0SGK43WsESf6GTaq3kwBvdV7QJYsandfuWKJ8z/Wu3Y/Eedh7s512ncQ0TFyatenknqGwAQBANChyAAAAAAikumRUq/0ymp3XrMAR9PorHdt/TJvv2azp3dNK70nrjk/eoelPTGvrx7Zq0z2bNDk3qQ0zGzSeGtfE9ITi6+J6Yd8LbfXRbe3sWdAq2dqPZYpq+x208a3sb1DHGdQgJsbDxnTYP4sorYX3CABAt7BcFQAAAICWqpuBVwsQtYWK2sJFq/Nq/1t7PEi7lW0kfw+O5NakprZPacv9W5TandLk3KTKdln5K3nFxmIyDEMVt6KyXZZruXIKjqxbll7+wss6+I2DnQenAyQ2h0eYGTfd6L9f/Q1icQUAAECiyAEAAAAgoEYzMFa+HvS8sNevde7UOc3um1Xy9qQmZiY0OTepjbMbZS1YKttlOQVHdtZWYjzhP5IJxZNxjW0a01sn3upakSNIQrrfSfMwKMq8L4oiQLubiPfzXhmEMQTRr2LQoMcFAIDVjCIHAAAAgKE0f3Ze8fVxJcYTiq+Py4ybKttlWQuWXMuVV1maHTJiyIyZMuOmzISpWCLmP8ZiuvzGZaUeSHU0jmZJ1TCbVw+DQRj3haf92NfbtLmbie5G1+4k2R12g/tu6WU8u6Vb8aoXi1Z7hPTjPgUAYC1hTw4AAAAATc3Pz3/gUe/1v/3bv11+/cyZMz15njmbkQzJMA15FU9O3lH+Sl7ZTFaFawXZOVuu5ariVuRVPMlbWvrKlIwRv03m9UzouHRiGJObg/yL9WoSufrohepm1GE3+A4az17fK602yB7ke7df92g7M3J6fZ8CALAWMJMDAAAAQENHjx7VvffeqwMHDnzoWO0yUv16Lk+qOBU5OUfFG0XFxmJyCo4Mw5BruSrdKsnJO3ItV2Wn7Bc8yp5f9IhIo4Rqq19vD2KxoJFBHXOjjam7vWF1o0JAo898pXYLHIMQ9+r7G1S9ilW967f63Pt1nwIAsFZQ5AAAAADQ1DvvvKPHH3/8Q6+n0+m6z2sLIt18nt7j92nnbBUXihq9OirDMGRnbRkj/mbjTt5R6VZJdt6WU/hgscMwDaX3vuEjQFwAACAASURBVD/uqLVKejdLbA5ScnuQxtLIII9tpTDxHJZ7pV/6HYOgBa61/BkBANBNFDkAAAAADKXp3dNyCo6sBUuFawXFRmOquBUlxhMyY6a8iifXcmXnbZUWS7JzS4WOgivX8h+pXZ3tx7Ha9Tt53IlBHPsgjmnYDXtMh338AAAMAoocAAAAAIbWjkd36Pzp835hY8TfeDwxnpAZNyVPKjtlOQVHds4vdFRndViLlnY+trMvY26WzGyW8Aw6MySqZGm71+v1+MIKO85Ol2sK8/7D3ivd0Oy9dxrTsPfMoNxTAACgv9h4HAAAAMDQOnzisJyCo+x8VrnLOWUzWWUvZZXN+H/n38urcLWg4vWiijeKKi2WZC1aUkU6+PWDXR1bN9fZX3ntqPvqNHnc7fG1um7YTb3b2UC6nT6HJRnfKp5S72Laql0vYxp0LO0eH5b7AgCAQcdMDgAAAABD7cirR3T8kePKXszKKTiKJ+OKJWKSKXllT2WnLLfgLs/g8DxPT732VGT9t0pwRpnArJ1NUK/fKPqqvW6rX+/Xey3M+IIk1xu1D1NACTvOVu3q6SSenQgb07AFqW7FtNVn3m5MuxmXIDNvAABA9JjJAQAAAGCopfem9cQrTyh3LaeFHy74szkyWeXmc8uzOfJX88rOZ+XkHD15+kmldnd/L44LT3fnF9rNksWDoJfja9ZXq/7CjrOTPgddq02z+xHTQRBk/GE2HF8N9wwAAIOAmRwAAAAAht7MgzN6pvSMTj12Sm9/823FxmLyKp4kyTANuZarnY/tDLxEVZDEYy+T9r3oO8prt3uNTvrsR9uo9tXoRNjCQafX7Vb7XsV02OICAABao8gBAAAAYNU49NIhHXrpkC6/cVmZ1zOS/JkeqV3dn7kBAAAAoPcocgAAAABYdVIPpJR6gMIGAAAAsNpR5AAAAACAVaydDY9ZUgcAAADDho3HAQAAAAAAAADAUGImBwAAAACsYszOAAAAwGrGTA4AAAAAAAAAADCUKHIAAAAAAAAAAIChRJEDAAAAAAAAAAAMJYocAAAAAAAAAABgKFHkAAAAAAAAAAAAQ4kiBwAAAAAAAAAAGEoUOQAAAAAAAAAAwFCiyAEAAAAAAAAAAIYSRQ4AAAAAAAAAADCUKHIAAAAAAAAAAIChRJEDAAAAAAAAAAAMJYocAAAAAAAAAABgKFHkAAAAAAAAAAAAQ4kiBwAAAAAAAAAAGEoUOQAAAAAAAAAAwFCiyAEAAAAAAAAAAIYSRQ4AAAAAAAAAADCUKHIAAAAAAAAAAIChRJEDAAAAADCwDMNYfnR6XthjAAAAGFwUOQAAAAAAA6laePA87wN/hzmPAgcAAMDqFOv3AAAAAAAAaKRauPA8r2kxotl5hmE0bN/sWD3zZ+eVOZuRJKX3pDW9e7rueXPPB7rch1x4unXbC0+3d83a67Vqu7LvdvsK02e716sV9Nr12gdpGzYeUfXXzT47jWmja0V9j3X6fejVZxH1PdrO5xDF9zbq7ywA9BJFDgAAAADAqlUtYrR7bKWTh0/q3Klziq+PS4akpWZOwdGOR3fo8InDEY24tbnnO0t8d3Jer6/V6nrVY2GSz83iGPY9tBpr1OMM22enMQ16rTDnRSnqmHYjnp3EJaqY9uOzAYAoUeQAAAAAAKxKURQ4Lr52Ucf2H1NyS1Kz+2aVGE/IMA1VnIrsnC1rwdL50+f13OhzOvLqEaX3piW1TmgGSSA3S6a2U+hoR/Wag5b0bBXPIMnn2uNB4xg2Hiuv2e1x1utz5XWDnN9un2FEFdNWwsY0bLtO49nJd29Qv7cA0CvsyQEAAAAAWLVWbkheuyxVs2OSX+B48XMvanr3tLY9vE0zD84ovSet1K6Utn50qzbfu1mT2yY1tX1KyduTOv7IcV363qWuvp8wS9gELahEuaxUlMnxIInhdq8RpGAQNqneTgK81XlBl2JqdF67rwfps53+V163H8sgtRvTMO06iWcncYkipt34zgJAr1HkAAAAAAAMrEYFiCDneZ73gUf1tVbHqo7tP6bN92zW9O5ppfekdccn79D0J6a19WNbtemeTZqcm9SGmQ0aT41rYnpC8XVxvbDvhWjeeEQGIancL0ELAL3+9fvK/gZ1nEENyuddK2xMh/2zCGsQP0MAaAfLVQEAAAAABlJ1Q/Bq4aK2CFG73FSz88I6efikkluTmto+pS33b1Fqd0qTc5Mq22Xlr+QVG4vJMAxV3IrKdlmu5copOLJuWXr5Cy/r4DcOdjyGTgzS/gfojn7/Ar/fxSES853jOwtgtaDIAQAAAAAYWI0KFitfD1LYaHbOymPnTp3T7L5ZJW9PamJmQpNzk9o4u1HWgqWyXZZTcGRnbSXGE/4jmVA8GdfYpjG9deKtrhU5giQl+5H87lfCnSTt+6IqAgzqPTaIYwhiUO/RYYkfAARBkQMAAAAAgBrzZ+cVXx9XYjyh+Pq4zLipsl2WtWDJtVx5laWZIyOGzJgpM27KTJiKJWL+Yyymy29cVuqBVEfjCLNpdNDj3dDPZOkgJGovPO1/ZvU2mO5morvRtTtNYg/SPdbLeHbLINyjKw3imAAgDIocAAAAAICBMz8//4G/p6enO3r9Rz/6kX7yJ39SknTmzBnt37+/4fPM2YxkSIZpyKt4cvKO8lfyKttleRVPds6Wa7mquBV5FU/ylvYCMSVjxG+TeT3TcZEjjLW0TNUg/xK9HzGJohAQJKa9fm+NNnKvjqNeUWlQDOo9OoxFIgBohiIHAAAAAGCgHD16VPfee68OHDjwoWNBl69q9nrta42ey5MqTkVOzlHxRlGxsZicgiPDMORarkq3SnLyjlzLVdkp+wWPsucXPSLSKDHaarbAWlqmatCSx402pu72htXNCgFBiwDtFDgGIe61hY5BNEixqjWo4wKATlDkAAAAAAAMnHfeeUePP/74h15Pp9N1z2/1eu3x2uJJvefpPf65ds5WcaGo0aujMgxDdtaWMeJvNu7kHZVulWTnbTmFDxY7DNNQem/98UShVfK6WeK3WwnOXvY5DEnaQR5bPe3GtB/32DAZhhjwGQJYTShyAAAAAABQY3r3tJyCI2vBUuFaQbHRmCpuRYnxhMyYKa/iybVc2XlbpcWS7NxSoaPgyrX8R2pX75eqWguGOfk6qGMf1HENK+IJAL1HkQMAAAAAgBV2PLpD50+f9wsbI/7G44nxhMy4KXlS2SnLKTiyc36hozqrw1q0tPOxnX0Zcz+WGepln+1eL+iMl34no8OOM4rlmsLENKprdarZe+80pmHvmUG5p5oZpM8QAKJi9nsAAAAAAAAMmsMnDsspOMrOZ5W7nFM2k1X2UlbZjP93/r28ClcLKl4vqnijqNJiSdaiJVWkg18/2NWxDfI+BN3SafJ1Zcy6FcNG1w06/rDjrHfeakjIt4qn1LuYtmo3DPEEgNWKmRwAAAAAANRx5NUjOv7IcWUvZuUUHMWTccUSMcmUvLKnslOWW3CXZ3B4nqenXnsqsv5bJVWjTKYGSSZH3WdQtWNo9ev9eq9V29Rr26oA0Or1dvdEaSTsOFu1a6STmIYVNqZhC1Ldimmrz7zdeHbjXhuE7y0A9BIzOQAAAAAAqCO9N60nXnlCuWs5LfxwwZ/NkckqN59bns2Rv5pXdj4rJ+foydNPKrW7+3txXHiapGU7miWze9lXq/7CjrOTPgdds/H3K6YAgMHDTA4AAAAAABqYeXBGz5Se0anHTuntb76t2FhMXsWTJBmmIddytfOxnYGXqAqSJO1l8r2fffbyOu1eo5M++9G2l+8v7DV79d760e+w3S+9uh4A9ApFDgAAAAAAWjj00iEdeumQLr9xWZnXM5L8mR6pXd2fuQEAAIDGKHIAAAAAABBQ6oGUUg9Q2AAAABgUFDkAAAAAAEDPtLORNMvnAACAVth4HAAAAAAAAAAADCVmcgAAAAAAgJ5hdgYAAIgSMzkAAAAAAAAAAMBQosgBAAAAAAAAAACGEkUOAAAAAAAAAAAwlChyAAAAAAAAAACAoUSRAwAAAAAAAAAADCWKHAAAAAAAAAAAYChR5AAAAAAAAAAAAEMp1u8BAAAAAAA6YxjG8nPP89o+r532jdq1agsAAAB0AzM5AAAAAGCIVQsN1QLDysJDkPM8z2tZoGh03WpbChwAAADoB4ocAAAAADDkqgWGVoWGoOettHIGBwAAADAoWK4KAAAAANBQqwJH0KWubp6/qe//zvf17rff1Y3zNyRJm+/ZrG2f3aYH/7sHNXnnZHSDBgAAwJpBkQMAAAAAUFerAsfKY43OP/3rp/VXf/BXkieNTY5p052bVClXVHivoLP/x1m98cIb2vOP9+ihf/FQ5O8BAAAAqxtFDgAAAABAQyv34mh36ao/fPgPNf9X80rtSmn89nElkglJkmM5Kt0qybphqXCtoO/+79/Ve2+8p8f/3eORjh8AAACrG0UOAAAAABhy1cJDo83B2z2vKuhMjUaOP3Jcl//6suYOzGnq3imNT48rNhZT2S6rtFhS4XpBhYmCRsZGZMQMXXj1gl78hRelvYG7AAAAwBpHkQMAAAAAhli1aFEtXNQWIWqLEq3OW/k8SDFjZbGkts3fnvpbXfzLi5r9zKxu+/htun3n7dpwxwbJkKwFS/krecXGYjJMQ17FU8WpqGyX9cPv/FDaIGl7e3EAAADA2kSRAwAAAACGXKOCxMrXg54XtJ9m7b79zLe1adsmbZzdqM13b9bWj2/V1PYpFa8Xlc1k5VU8le2ynKIjp+DIztuyc7ZKyZKcbzkUOQAAABCI2e8BAAAAAABWl9x8Tjd+cEPrb1uvdZvWaXRydHkvDjNmamR0RLGx2Icf62KKJ+PSDcnIB1tSCwAAAGsbMzkAAAAAAJHKnM0sFzOMEUMVtyJrwZIMqeJW5FquKm5F8vwlrwzTkDliyoz5D8M05GWC7/0BAACAtYuZHAAAAAAwhM6cOaOzZ89+4FF14cIFHT16dPnvXj9f+MGCzBHTX5Kq5G8ynrucUzaTVeFKQaWFkpy8I9dyVXbK8sqevIq3XPQYMUakG6HCMlSqe6QE2TC+0XmtrtHoeNC+AQAABh0zOQAAAABgCH3nO9+R53n60pe+9KFjc3NzevbZZ5f/7vXzd/6vd/wZG0VXpVslFa4VZI6YsnO2zBFTZbssO+fvweEU3i92VNyKKm5F5UpZ2txONIZP7Qbv1WJDvT1Omp23sk2rv9s9DwAAYBhQ5AAAAAAARCr9ibQq5YpKt0oq3igqvj4ur+IpkU3IjJnyyp6coiM7a/uPvC234PrFDrssr+LJS6/+pHu1sFAtYLR7XrPCRNSFi/mz88qczUiS0nvSmt49Xfe8uefDXf/C063bXni68bFO2ja7RpB2QTQaX9Drhx3byna97q9bfXYaz2bXa3WNdmPa6b3Zq88i6nu0m3GJ4vsOYHWhyAEAAAAAiNT49Lim7p5S8VpR+Y15mTFTZaesxHhCI/ERfxkruyw7b8u+tfRYmtXhFB1pi6Rkv9/F8GtUEGmnWHLy8EmdO3VO8fVxyZC0dKpTcLTj0R06fOJw5ONuZO757iQvmyVMo+iz1fWlcAndZmMLW2zqJBZhxhmmz07j2U7/Yc6LUtQxbdSuk5j2Iy4AsBJFDgAAAABA5H76N39apx4/pfhEXIZhyC25y0UOSao4FTmWIztnq3SrJOuWJTtnyy260uf7PPghVG/mRqMlqYIsV3XxtYs6tv+YkluSmt03q8R4QoZpqOJUZOdsWQuWzp8+r+dGn9ORV48ovTctqXUiNEjyuVkStlVyt5OCRLvJ306vXXv9IMnn2uPtxqPd99GosNCtcdbrc+V1W53bbn9hRRXTVsLGNGy7TmPaq7hE1RbA6sLG4wAAAACAyN339+7Tnfvv1OKFRWXns8pmsspeWvrvfFa593IqXC2oeL0o66al0kJJhesF3fXwXdK9/R79cIl6aaqLr13Ui597UdO7p7Xt4W2aeXBG6T1ppXaltPWjW7X53s2a3Dapqe1TSt6e1PFHjuvS9y5F1n893Upmtiq+RNFvFEWZlecFKRiETaq3kwBvdV7QJYvaiX+URa52i2/9SKq3G9Mw7TqJab/iAgC1KHIAAAAAALri8T95XLOfntXijxaVy+T8Akcmq9x8Trn3csq/l1f+al6FawXlr+V190N36wv/5xf6PeyeqS4Z1Ww/jlbndWPT8GP7j2nzPZs1vXta6T1p3fHJOzT9iWlt/dhWbbpnkybnJrVhZoPGU+OamJ5QfF1cL+x7IdIxIHgBoNfLBa3sb1DHGdQgJujDxnTYPwsACIvlqgAAAAAAXfPEv3tC3/pn39LrX3ldufdyGhkdUWzU/7+iruWqXCprZN2IPvXrn9KBowf6O9gequ6LUS1cNFo+qtV5tf+tPd5s341mx04ePqnk1qSmtk9py/1blNqd0uTcpMp2WfkrecXGYjIMQxW3orJdlmu5cgqOrFuWXv7Cyzr4jYOdB6cD7Wx+HGb/BkSn3/Hvd3GI+87XSVyIKYAqihwAAAAAgK566MsPac9/vUff/8r39e633tX1/3RdkrR5+2bd9dBdevBXH9SGOzb0eZS912gGRrO9NYK8HuR4o2PnTp3T7L5ZJW9PamJmQpNzk9o4u1HWgqWyXZZTcGRnbSXGE/4jmVA8GdfYpjG9deKtrhU5Ot1IO0jysx8JU35R/74o4t/uJuL9TIoPwhiC6FcxqJPPf9BjCiB6FDkAAAAAAF23cW6jHv6th/s9DDQxf3Ze8fVxJcYTiq+Py4ybKttlWQuWXMuVV1maVTJiyIyZMuOmzISpWCLmP8ZiuvzGZaUeSHU0jmZJ1Xb2Dmg34d3o9V4kTAchKXvhaf8919tgupuJ7m7EP+wG993Sy3h2Szfi1UlcVkNMAUSHPTkAAAAAAOih+fn5Dzxavf7mm28uPz9z5kzXnmfOZiRDMkxDXsWTk3eUv5JXNpNV4VpBds6Wa7mquBV5FU/ylpbKMiVjxG+TeT0TOi6daLVBdZAEaHUD5V5tpDzIvzqvFjuqj17oNP5B49nrZHgU92a/dPMe7SQuwxxTAN1BkQMAAAAAgB45evSovv3tb8vzvOVHVe1rK1/v1XN5UsWpyMk5Kt4oKnfZ3zA+/15exetFlW6V5OQdfz8Vp+wXPMqeX/SIyMpkd23Su93kZdDkbLOkaTcSpoNa4GhUYOh24afT+Ldb4BiEuA/CGJrpV6w66W/QYwqge1iuCgAAAACAHnrnnXf0+OOPf+j1dDpd9/xdu3YtPz9w4EDXnqf3+P3bOVvFhaJGr47KMAzZWVvGiL/ZuJN3VLpVkp235RQ+WOwwTEPpvfXfQxSaLac0TAYp0d7III9tpTDxbFY4GYbPp9uIAYBhQ5EDAAAAAABoeve0nIIja8FS4VpBsdGYKm5FifGEzJgpr+LJtVzZeVulxZLs3FKho+DKtfxHaldn+3GsdsOcPB7EsQ/imIYdMQUwjFiuCgAAAAAASJJ2PLrD34PjamF5qapsJqvsfFa5yznlr+ZVuFZQ8WZRpcXS8qwOa9HSzsd29nv4HxJ2E/NuaDd53Gq5pkFJRocdZ6fjDvP+Gy2FVnuNXu/J0miczc5pFdOw98wg3FOdLA/HXhzA2kWRAwAAAAAASJIOnzgsp+AsFzWymayyl/xCR+5yTvn38ipcLah4vajiDb/QYS1aUkU6+PWDXR1bq8Rts9fDbEYdZcK302utHF+3krmdFlTCjrPd+A9CMj6IbtybYWPaql0vY9pJXKKIKYDVh+WqAAAAAADAsiOvHtHxR44rezErp+AonowrlohJpuSVPZWdstyCuzyDw/M8PfXaU5H13yoZ2yzp3a7qPh+dXKOV2uu2O7Ok1fg6SQQ3ah8mDmHHGSb+ncSzE2Fj2q17M2xMW33m7ca013HptC2A1YmZHAAAAECEDMNYfoQ9r9Gx2tebHQOATqT3pvXEK08ody2nhR8uLC9ZlZvPLc/myF/NKzuflZNz9OTpJ5Xa3f29OBotI9QsmR1k6aFmyeJB+EV4s/H1sq9O4titPgddN+/NbrTrlU7i0mlMAaxOzOQAAAAAIlItMHiet1xw8DyvrfNWtln5d73r1b5OkQNAFGYenNEzpWd06rFTevubbys2FpNXWfp3xjTkWq52PrYz8BJVQRKPnSQnO01sdjMxGsW1271Gv2IZtm27+2p0Q9jCQafX7Vb7XsW0H3GhkAFgJYocAAAAQISqxYZqAaPd8xoVMbrh5vmbypzNSJLSe9LadNemnvUNYDgceumQDr10SJffuKzM60v/XuxNK7Wr+zM3AAAAgqDIAQAAAAyRKAoif/wrf6y//sO/lkxpJDYiSSo7ZcmTfuLJn9Av/N4vRDJWAKtH6oGUUg9Q2AAAAIOHIgcAAAAwoFotVdVoOaxGrrx9Rb//U7+vxLqEbvsvbtO6TetkmIbKTllOzlHxRlF/842/0dvfeFv/6Lv/SFvu3xLZewHCClrYa3Req/ZB2rXqG2tLO5ses6wOAADdR5EDAAAAGEDtFjBaufo3V/W1z35NW+7eoqmPTmk8Na74+ri8iic7b8tasFS8VtToxlFlM1kd23dM//A//MPI+gfCiGKfm2b71YTdAwcAAACDgyIHAAAAEKFqkrTVBuDNzou6wCFJX937VW25f4um90xry/1bNJGe0EhiRE7Bn8FRuFpQbCwmI+aP59bFW/rqnq9Kvx7pMIC2dbrPTZBrR2X+7PwH9rmZ3j0d6fUxGJidAQDAYKHIAQAAAESk9lfk1b+rVv6yvNl5tf+tPd5s+ZzaYyuv+/Lff1mJiYS2fHSLtn50q1IPpLRxbqMqdkX5a3nF1sVkmIYq5YrKdlmu5coturKztvRHkv6rzuICDLJmy1kFXSrr5OGTOnfqnOLr45IhaelUp+Box6M7dPjE4UjHDAAAgPdR5AAAAAAi1CgRuvL1oOdFceytF9/S7E/Najw1romZCW3atkkb5zbKummp4lbkFlw5OUf2hK3SeEmJ8YTi43GNTY7JftOmyIFVrVkxsla9GVYXX7uoY/uPKbklqdl9s0qMJ/yCoVORnfOXgTt/+ryeG31OR149ovTedPffEAAAwBpDkQMAAABYxd578z3F1sWU2JBQPBlXbDSmsltWabEk13blyZNhGjJGDJkxUyOJkeVHbCwmxSTjSrClf4C15OJrF/Xi517U9O5pTW2fUvL25PI+N07OUXGhqMK1ghLjCWXnszr+yHE98coT/R42AADAqmP2ewAAAADAanDlypUPPJq9funSpeXj3/3ud7v6/NUTr/pLY40YfvK14KhwtaBsJqvi9aLsnC235KpSrsireJLn/2J9ufBhmPIusfky+qfeEm6dnFevTRjH9h/T5ns2a3r3tNJ70rrjk3do+hPT2vqxrdp0zyZNzk1qw8wGfwbV9ITi6+J6Yd8LofsDAABAfczkAAAAADr0W7/1W9q+fbv27NnzoWOlUulDrxWLxeXnN2/e7OrzfDYvw/CXz3Hy/ibjsbGYnLwjGZJbclW6VZKTd+SWXJWdsiquX/DwKp4MGTIqzORAf0S5z03t80ablDdqt/LYycMnldya1NT2KW25f4tSu1OanJtU2S4rfyWv2FjM/9657+9z4xQcWbcs2SdtiS06AAAAIkORAwAAAOiQZVl688039fnPf/5Dxz7ykY986LV77rln+fnP/dzPdfX5Q3//IX3tD74mO+/vD5C/mpcMyc7ZMkaWih8F5/1CR9H1ix22X+yoeBV508zkQP90c5+bsO3OnTqn2X2zSt6e1MTMhCbnJrVxdqOsBUtluyyn4MjO2kqMJ/xH0l8ubmzTmOxzdtPxAAAAoD0UOQAAAIBVLL0nLafoyFqwVLheUGwsJq/syc7aMmOmKuWKyqWyv0nyoiU7Z3+g2OE5nsReycCy+bPziq+PKzGeUHx9XGbcVNkuy1qw5FquPwPKeH+fGzNuykyYiiViiiWW9rm5zOwoAACAqLAnBwAAALCKGaah+/7efcpmsspfySt3OadsJqtbl275r72XV/5KXoVrBVk3reVCh523VVosSR/r9zvAWnXfffdpfn5++VF19uzZ5ednzpzp+fPM2Yxk+N8tr+LJyTvKX8krm8mqcK3g73NjucvLvlX3uZGp5X1ulGk/HgAAAKiPmRwAAADAKveFP/qCjppHlb+SlyS5lqt4Mq6RxIgk+fsGlMqy87bsrL+slb3ob0iuX5TEalXosWeffVaZTKbuklG1r/XruTz5S73lava5KTgyDEOuVbPPjVWzz03Z3+cGAAAA0aLIAQAAAKwB/+DP/oFO/uJJeWVPbvH9IodhGvLKnr+PQNGRk3NUypZUcSv64p9/UV8787V+Dx1rVDpdf520PXv2LD8/cOBAz5+n9/jjsnO2igtFjV4dlWEYsrNL+9y4FTl5f58bO2/LKawodnievDTFDgAAgKhQ5AAAAADWgLt+5i794olf1L/92X8ra8HS2OSYRkZHZJqmPM9TxanILbmyFi05BUe//O1f1tz+OelM62sDa8n07mk5haV9bq4VFBuNqeJWlBhPyIyZ8iqeXMtdXvLNzi0VOgquv2eH60mpfr8LAACA1YMiBwAAALBGbPuZbXrWe1Zf/7mv6wff+sFyQlby9xeouBXd/TN36/E/fbzPIwUG245Hd+j86fN+YWPE33g8MZ6QGTclTyo7ZTkFR3bOL3RUZ3VYi5b08X6PHgAAYHWhyAEAAACsMU+88oTsnK3M9zP68V/+WJL0kb/zEaX3pJUYT/R5dMDgO3zisJ4bfU7Z+awkf5+bRDIhM2HKMPyCoWu5cgr+slWlxZJf4KhIOiT2uQEAAIgQRQ4AAABgDUqMJ3TnZ+/UnZ+9s88jAYbTkVeP6Pgjx5W9mJVTcBRPxhVLxCRT/j43TlluwV2eweF5np567Sn9Rdv6GwAAIABJREFU3h//Xr+HDgAAsKqY/R4AAAAAAADDJr03rSdeeUK5azkt/HBB2UxW2UxWufmccpdzyr+XV/5qXtn5rJycoydPP6nUbjbjAAAAiBozOQAAAAAACGHmwRk9U3pGpx47pbe/+bZiY7EP7HPjWq52PrZTB79+sM8jBQAAWL0ocgAAAAAA0IFDLx3SoZcO6fIbl5V5PSPJn+mR2sXMDQAAgG6jyAEAAAAAQARSD6SUeoDCBgAAQC9R5AAAAAAAoA8Mw1h+7nleqPOCXMMwjA8cq20TpH8AAIBBxsbjAAAAAAD0WLXQUC0u1Cs8tDqvWryoPupdo1FBo/YBAAAwzJjJAQAAMKA6/YVvq/ad/jIYANCZ6r+vjQoUrc5r9e9ztQjS7NorZ3k0Mn92XpmzS/uN7Elrevd03fPmnm95qbouPN267YWnGx9r1LZZm1btg7YNe/12+gg7vpXtet1ft/rsNJ7NrtfqGu3GtJP7ulH7bnwWUd+jYePSaTyDXgfA6kKRAwAAYADV/nLXMIyGSahm5zX7dXC9pUtqz290DAAwWJoVuqP4t/vk4ZM6d+qc4uvjkiFp6ZJOwdGOR3fo8InDHfcR1Nzz9ROXzZKe1WPtJsuDtA2q0z4atW8Uj1Z9NtNqrFGPM0yfUX9mQWMVNqadiDqmvf4OtbpmmP4AoB6KHAAAAAOq01/4Brl2u8cAAIOlXmEjaIGj2XkXX7uoY/uPKbklqdl9s0qMJ2SYhipORXbOlrVg6fzp83pu9DkdefWI0nvTklonQoMkLpslYZsld9tt1+rX+0H6DKpVXIIkn2uPBx1b9Vi7iehGserWOOv1ufK6rc5tt7+wooppK2FjGrZdpzENE5cw37sgxR0KJMDaw54cAAAAa1R15ke9JFezY7Wu/8fr+pP/9k/0lR1f0ZfXf1lfXv9lfeXjX9Gf/uqf6sZ/vtHttwBEpvaeD3Neq/ZBrw9EZeU91869d/G1i3rxcy9qeve0tj28TTMPzii9J63UrpS2fnSrNt+7WZPbJjW1fUrJ25M6/shxXfrepW69FUmtk6vtFD6CnhNVojRIYrjdawQpGIRNqrebWG52XtClh9r5DKKIZ9D+V167H8nzdmMapl0nMQ0Tl06/swCwEkUOAACANarZZrWtNrKVpFf++1f0Bz/1B/qbb/yNXMvV1PYpbb57s0oLJb35tTf11b1f1el/croXbwXoSBQbQDfbwDnI5tBYm4IWIBqd16hdvY3F25mld2z/MW2+Z7Omd08rvSetOz55h6Y/Ma2tH9uqTfds0uTcpDbMbNB4alwT0xOKr4vrhX0vBL4+gglaAOj1Mkor+xvUcQY1iEn1sDEd9s+iE6vxPQEIjuWqAAAA0LZ/s//f6MpbV5R6IKXk7UklkglJkmu5Kt0qqXijqMK1gr7/u9/X/Jvz+uLpL/Z5xEBz3VweLqzr71yXJE1tn+pqP+iP2r2Uqn9XrdxfqdF57W5EXqvRTL2Th08quTWpqe1T2nL/FqV2pzQ5N6myXVb+Sl6xsZgMw1DFrahsl+VarpyCI+uWpZe/8LIOfuNge4HAmtXvpYX6XRwaxOLKakBcgbWJIgcAAMCAql1bPYrz6rVp95gk/eHDf6hr/9813fnZO7X53s2amJ5QbCymsl2WtWipeL2oxHhCI6MjMkdMXfx/L+rrP/d16acCDw9Ys9746ht67bdf043/fEOxhP9/11zb1dQ9U/o7/+TvaNeRXX0eIaLU6N/ala93uo9SvXMatTt36pxm980qeXtSEzMTmpyb1MbZjbIWLJXtspyCIztrKzGe8B/JhOLJuMY2jemtE291rcjR6UbajZY9mnu+9d4G3Uya8uvz90VRBGh3E/F+JsQHYQxB9KsYNOhxATBYKHIAAAAMoCh+4Vtb9Fh5vNmvf5sdO3fynC5975Lm9s/pto/fptt23qYNd2yQJFkLlgpXC4qNxmSYhryKt/xL3x/9hx9JmyTd11lcgGEUpAjpllz9/oO/r8WLi1q3aZ1m9szITJjLmzznr+b1Z//jn+l7v/M9/cr3fkXGCEteIXrzZ+cVXx9XYjyh+Pq4zLjpF7AXLLmWK6+y9L85I4bMmCkzbspMmIolYv5jLKbLb1xW6oFUR+NollQNusdD0Da1hY4w/UVlEBK6zYo+3Ux0N7p2J8nuTvZi6YZexrNbuhWvdr+zza4xCN8jAP3BnhwAAAADauV66rWvBz2v3rrsrdo1O/ad/+U72nT3Jm2c3ahNd23SbTtu08yDM9p01yaNT49r3ZZ1Gts8ptGNo/5jw6gSG/xf++pbnUQDGG7Nvm+lbEn/ctu/VGmxpLlPz+muh+7SzCdnlNqV0m0fv01b7t+i2z52mzbeuVE3372p3579bdl5uw/vAlG57777ND8/v/yoqn2t0etXrlzRmTNnlo9F+TxzNiMZWi5UO3lH+St5ZTNZFa4VZOdsuZariluRV/Ekb6mIbkrGiN8m83omgghFa1ATyoOcmK0WO5oVgKJW3Yw67AbfQePZ6/uh1ebpg3p/Sv27Rwc5JgAGEzM5AAAAEEg2k9XNd29qdt+sxjaNaXRyVPFkXF7FkxkzNZIYUWw05j/GVjySMenHknL9fhdAfd1cHq5e+1r/+uP/WvFkXHd88g5N3TelifSERhIjci3XnyF1reD/qj5hyjAMLf54Ub+783elJ0MNAX327LPPKpPJtLWEVLNid+TPPaniVOTkHBVvFBUbi8kpODIMY3nfJSfvyLVclZ2yX/Aoe37RIyKNEqrNlpZq1jbsklRB+gtrUAscjTam7vaG1WGWE6vVboFjEOJefX+Dqlex6vV3D8DqRJEDAABgwIyMjNRddqpcLmtkZKRvzzOvZ2TEDI2Mjsgw/U1nrZuWJKniVpZ/3Vv9Za9hGjJHTH9Jk5gp0zRVzpS7FDUgvG4vD9dsY+i/+I2/UOlWSXc9fJe27tiq1E+ktOEjG+RVPBWvF5W7kpMZM/3Ec3Wj55KrxQuL0nck7Y8uDuiddDrd0eu33Xbb8vMDBw5E9nx+gz97xM7ZKi4UNXp1VIZhyM7aMkb8f/edvKPSrZLsvC2n8MFih2EaSu+t/x6i0E7SO0i7VkncsP21MkiJ9kYGeWwrhYlns+LCMHw+3dbvGHSjwAVgdWO5KgAAgAHy8MMP6+d//uf1xhtvLD+qfud3fmf5+W/+5m/2/PnCDxdkjpjyKp7KpbJKCyXl3sspm8kqfyUv66YlO28vJ7yWf9lbU/TQzfZjAvRCN5eHa9Sm4lb0+u++rqntU8sbPE/dN6XbPu7vdbN+63qtm1ynsY1jGt0wqtGJ0eXNnkc3jkrfkwyPvTkQnend03IKzvIMotxl/9/4bCar3HxO+ffyKlwrqHizqNJiSXZuqdBRcOVa/iO1q7P9OFa7YU7IDuLYB3FMw46YAhhGzOQAAAAYIJ/61KcaHvu1X/u15ee/8Ru/0fPnm+/e7M/YKLqyFv0EmBkzZef8X/iWS2XZOVulbOlDv+6tuBVVyhUZUyRkgaqbP7gpt+D6y79NjCq+Pi7DMOQUHH+DZ9PwN3aO+8vBjYy+vyRcfF1cuiZ5N6NbIgiQpB2P7tD50+eVGE/IHPE3Hk+MJ2TG/RlFZacsp+D4/94vlpZndViLlnY+trPfwx9o7SaPW/2afVCS0WHH2elyTWHef7Nzex3PZu+905iGvWcG5Z4KapCX+wLQW8zkAAAAQCDpPWlV3IpKt0oq3ij6m9HO1/zC90pexetFWQvWh5cyscv+rI7urWIChJLNZj/wWPm6ZVm6cOHC8utRPp8/Oy8Z0kjcXxrOLbkqXC8om8mqeL0oJ+8sf3c8z1veENoYMfwl4AxTGrw9njHkDp84LKfgKDuffX8mx6Wlf+svL83muFpQ8XpRxRv+jA5r0ZIq0sGvH+zq2BolNFslOsMejzKB2mnyeOVYupXcbRWLdjf1DjrOeue12i8lyHj6Lci91auYBv0e9CKmnX5nVxr0+wBA9zGTAwAAAIEkb09qavuUCtcKGt0wqpH4iMpOWYnk+7/wdUuunIK/XnvpVklOzpFT8B/aInnr+dU5BsfXvvY1bdu2TanU+8vrTExMSJLm5/29CcbHx/Xtb39bv/zLvyxJkT7/WPZjy/vbOEVH1k1Lucu55U2enaIjO2vLLfqFworjz4hSxV8CyzRMVQqVrsUHa9eRV4/o+CPHlb2YlVNwFE/GFUvEJFPyyp7KTlluwV2eweF5np567anI+m+V4GyW9G6nXe1Mgla/qu9E7bXb7afVGFv9Ir/V62FjWW8cYcYZ9DNoNL5ufm7N+m32+sp+wxakuhXTVp95uzHtZlwoXgAIiiIHAAAAAvuZ3/oZnfjFE0qMJ2QYhtySq0Qy4f8S3fCXMXEt11/G5FZJ1i3LT9JarvR3+z164IN++MMf6t1339WXvvSlDx3bvn378vNqYSLq5xfOXJBX8WTnbZUWSspfzcswDdk5e3mZIDvvf5ec/PuzospOWZ7rqVwpy7udwiGil96b1hOvPKEX9r0g65alsU1jiiViMkYMeRXPX7rQcpdncDz12lNK7e7+XhxBkrrttKs91k4Sudcavb9ujK+TvsK27eX767Vm9+Zajmkn39mqYZnNA6A3KHIAAAAgsHv+y3t098N36/y3zsureHIsxy9yJPwih1f25JZcuQVXpZw/m6Nwo6Dtf3e7zt19TiIfCyyb/sS0ynZZpcWSCtcLiq2Lyat4SmQTMmOm/32yXJWyS/se5N5fAs4tufLKnowZ9rlBd8w8OKNnSs/o1GOn9PY331ZszL8/JX/ZNNdytfOxnYGXqAqa0A2r00RnNxOlUVy73Wv0K5Zh20a1r0YnghQOunHdbrXvVUyHLS4AVieKHAAAAGjLL/3RL+mbB7+pH/zfP5BTdJRYn9DI6IgM0/+Fb3U2h5P3l63a/gvb9eiJR3X06NF+Dx0YKInxhD7yUx/R9f90XaMTozJj5oeWgCvb/ibPpWxJpcWS7KwtO2/LztnSrOTFqByiuw69dEiHXjqky29cVuZ1fxOY9N60Uru6P3MDAAAgCIocAAAAaNsvvfxL+vf/67/XX/72Xyr/Xl4joyOKjcb8fTksV67jKjYW02d+4zP69D/9dL+HCwyszx//vP7V9n+l3JWcZOj9JeAS/mbk1WWBqstWVQsdruVKh/o8eKwpqQdSSj1AYQMAAAweihwAAAAI5TPPfEa7juzS2d87q3f//F1d+4/XJEm37bxNdz18l/b+471KppJ9HiUw2DbObtRnnv2MXv0Xr/pLwBU/vARc2S77m5Av7XVTypb001/+aZ3On2YJOKAP2tlImiV1AADoPoocAAAACG3DzAZ99p9/Vp/955/t91CAobXvn+5TfCyuv/hnfyE7a2t0YvT9JeA8TxVnaTZH1pZjOfrZ/+1ntfdX9+r00dP9HjoAAADQdxQ5AAAAAKDPPvk/fFKzn5rVyV86qdzlnMpuWSNxf8mqsl3WSGJEyS1JffHPv6jUbpYMAvqJ2RkAAAwWihwAAAAAMADSD6b19A+e1o/+nx/p3W+9qx+/9mNJ0kc+/RFt++w2zX56ts8jBAAAAAYPRQ4AAAAAGBSGNPvpWQoaAAAAQEBmvwcAAAAAAAAAAAAQBkUOAAAAAAAAAAAwlChyAAAAAAAAAACAoUSRAwAAAAAAAAAADCWKHAAAAAAAAAAAYChR5AAAAAAAAAAAAEOJIgcAAAAAAAAAABhKFDkAAAAAAAAAAMBQosgBAAAAAAAAAACGEkUOAAAAAAAAAAAwlChyAAAAAAAAAACAoUSRAwAAAAAAAAAADCWKHAAAAAAAAAAAYChR5AAAAAAAAAAAAEOJIgcAAAAAAAAAABhKFDkAAAAAAAAAAMBQivV7AAAAAAAAhGEYxvJzz/PaPq/29XrXaHb9oH0DAACgu5jJAQAAAAAYOtUiQ7XAsLJgEfQ8z/OWHyvb1R6rbdfsGAAAAHqLmRwAAAAAgKFULUy0KjQEPa9em3aPNTJ/dl6ZsxlJUnpPWtO7p+ueN/d825eWJF14unXbC0+3d83a67Vqu7LvIH01Gm+74+xmH/Xah3lvve6vm31G+bl18x7r9PvQq88i6nu0nc8hqphG+Z0FgDAocgAAAAAA1qxuLzt18vBJnTt1TvH1ccmQtNSFU3C049EdOnzicOR9NjL3fGeJ707Oa6dd9VinidNO+2jUvlkcuxWPqMcZts8oP7du32OdiDqm3YhnJ3EJ07YX31kACIsiBwAAAABgTWq0RNVKjV5vduziaxd1bP8xJbckNbtvVonxhAzTUMWpyM7ZshYsnT99Xs+NPqcjrx5Rem9aUuuEZpAkYrNkajuFjnZUr9lu8rTbY20VzyDJ59rjQccWVTy6Pc56fa68bpDz2+0zjCjvsWbCxjRsu07jGTYuYdv2498XAGiFPTkAAAAAAGggbIHjxc+9qOnd09r28DbNPDij9J60UrtS2vrRrdp872ZNbpvU1PYpJW9P6vgjx3Xpe5e6+j7CLGETtKASJqnZrF1USdIgieF2rxGkYBA2qd5OArzVeUGXHWr3M4gipkH6X3ndfiTO241pmHadxLOTuIRp24vvLACERZEDAAAAADCUqktNtdpnI+h59dq1W+CQpGP7j2nzPZs1vXta6T1p3fHJOzT9iWlt/dhWbbpnkybnJrVhZoPGU+OamJ5QfF1cL+x7oa2xddtaTloGLQD0ehmllf0N6jiDGsR7LGxMh/2zAIBhx3JVAAAAAIChU91EvFq4qC041BYgWp238porj9Xbs6PZsZOHTyq5Namp7VPacv8WpXanNDk3qbJdVv5KXrGxmAzDUMWtqGyX5VqunIIj65all7/wsg5+42CHkekMSdjVr9/7J/S7ODSIxRUAQGcocgAAAAAAhlKjmRQrXw96XhTHzp06p9l9s0rentTEzIQm5ya1cXajrAVLZbssp+DIztpKjCf8RzKheDKusU1jeuvEW10rcgRJLPc7+d3LcVDMeV9URYBhuccGYQxBDNM9OiwxBbB6UeQAAAAAACAC82fnFV8fV2I8ofj6uMy4qbJdlrVgybVceZWlWSUjhsyYKTNuykyYiiVi/mMspstvXFbqgVRH4wizaXTQ491Qb7y9GscgJGUvPO3HoN6mzd1MdDe6dqcJ60G6x3oZz24ZhHt0pX5+ZwGgHvbkAAAAAAAMlfn5+Q88on79zJkzoZ5nzmYkQzJMQ17Fk5N3lL+SVzaTVeFaQXbOlmu5qrgVeRVP8paWvDIlY8Rvk3k9E1GU2jNoyd9eJPcHMSlbLXZUH71Q3VC6k42sg8S01/dYq43cB+2erzXI92gjgxxPAKsfMzkAAAAAAEPj6NGjuvfee3XgwIEPHWt3Waogr7f7XJ5UcSpyco6KN4qKjcXkFBwZhiHXclW6VZKTd+RarspO2S94lD2/6BGRRonRVrMF+pVQrddvo7F2qt/vtZFGG1N3e8PqRoWAduLfToFjEOJefX+DapBi1Ugvv7MAEARFDgAAAADAUHnnnXf0+OOPf+j1dDpd9/x2X68toLTzPL3Hv56ds1VcKGr06qgMw5CdtWWM+JuNO3lHpVsl2XlbTuGDxQ7DNJTeW39MUWiVvG6W+O114rXdRHsQw5o8HmTtxnSQ7rFBNMwx6MZ3FgCCosgBAAAAAEAEpndPyyk4shYsFa4VFBuNqeJWlBhPyIyZ8iqeXMuVnbdVWizJzi0VOgquXMt/pHZ1th8H6hvm5PGgjn1QxzWsiCcAhEeRAwAAAACAiOx4dIfOnz7vFzZG/I3HE+MJmXFT8qSyU5ZTcGTn/EJHdVaHtWhp52M7+zLmYVlmKKx230PQGS/9jknYcUaxXFOYmEZ1rU41e++dxjTsPTMo9xQADCs2HgcAAAAAICKHTxyWU3CUnc8qdzmnbCar7KWsshn/7/x7eRWuFlS8XlTxRlGlxZKsRUuqSAe/frCrYxukfQhajSWqsXaaPF45jm7FsNF1g44/7DjrnbcaEvKt4in1LqZB7/VBjqfUu+8sAITBTA4AAAAAACJ05NUjOv7IcWUvZuUUHMWTccUSMcmUvLKnslOWW3CXZ3B4nqenXnsqsv5bJRujTKYGSSY36jNIUrSTsdZev9Wv9+u9Vm1Tr22rAkCr18PGo944woyzVbtGOolpWGFjGjbp3q2YtvrM241nN+61KGI66MUaAKsTMzkAAAAAAIhQem9aT7zyhHLXclr44YI/myOTVW4+tzybI381r+x8Vk7O0ZOnn1Rqd/f34rjw9OAkIFuNYxDG2iyZ3cu+gsSqndej6HPQNRt/v2I67IbhOwtg7WImBwAAAAAAEZt5cEbPlJ7RqcdO6e1vvq3YWExexZMkGaYh13K187GdgZeoCpI87GXyPYo+u50QjeL67V6jX/EI27aX7y/sNXv13vrR72q/XwCgVyhyAAAAAADQJYdeOqRDLx3S5TcuK/N6RpI/0yO1q/szNwAAANYCihwAAAAAAHRZ6oGUUg9Q2AAAAIgaRQ4AAAAAGHKGYSw/9zwv1HmNjtW+3up4s3bNxgUE0c5G0iyrAwDA2sHG4wAAAAAwxKrFhGoRoV5RotV5zY55nveBx8pr1h6rV9io1w4AAACICjM5AAAAAGDIVYsI9QoNQc8Lco1qUaNb5s/OK3N2ad+KPWlN757uWl8YPszOAAAA9VDkAAAAAACEsrIgEnQJrJVOHj6pc6fOKb4+LhmSlk51Co52PLpDh08cjnTcAAAAWD0ocgAAAAAAWqo3i2Pla7V/tzpXki6+dlHH9h9TcktSs/tmlRhPyDANVZyK7Jwta8HS+dPn9dzoczry6hGl96a79O4AAAAwrChyAAAAAAB67uJrF/Xi517U9O5pTW2fUvL2pOLr4/Iqnpyco+JCUYVrBSXGE8rOZ3X8keN64pUn+j1sAAAADBiKHAAAAAAw5KqzJJrtx9HqvKDXiMqx/cc0vXta07unteX+LZqYmZAZN+XkHRVvFDV6dVSx0ZjMEVOSlL2Y1Qv7XpD+554MDwAAAEOCIgcAAAAADLFqYaJanGi2fFSj85odW3mden3X/l3bZuW5VScPn1Rya1JT26e05f4tSu1OaXJuUmW7rPyVvGJjMRmGoYpbUdkuy7VcOQVH1i1L9klbYosOAAAALKHIAQAAAABDrtGm3itfb7b5d9THmrU5d+qcZvfNKnl7UhMzE5qcm9TG2Y2yFiyV7bKcgiM7aysxnvAfyYTiybjGNo3JPmc3vC4AAADWHrPfAwAAAAAArB3zZ+cVXx9XYjyh+Pq4zLipsl2WtWDJtVx5laVZJSOGzJgpM27KTJiKJWKKJWJSTDIu92ZJLQAAAAw+ihwAAAAAMKTuu+8+zc/Pa35+XouLizpz5szysUF9njmbkQzJMA1/k/G8o/yVvLKZrArXCrJztlzLVcWtyKt4kre09JUpv/BhmFImSHQAAACwFrBcFQAAAAAMoWeffVaZTOZDe2sMw3N5UsWpyMn5m4zHxmJyCo4Mw5BruSrdKsnJO3ItV2Wn7Bc8yp5f9AAAAABqUOQAAAAAgCGVTqc/8PeBAwcG/nl6jz9mO2eruFDU6NVRGYYhO2vLGPE3G3fyjkq3SrLztpzCimKH58lLU+wAAACAjyIHAAAAgEAM4/19EJptKt3ovFbtw7bDcJnePS2n4MhasFS4VlBsNKaKW1FiPCEzZsqreHItV3beVmmxJDu3VOgouP6eHa4npfr9LgAAADAoKHIAAAAAaKlaaPC8pU2hDaNpoaLeedX/1hYtatutLGwEaYfhtOPRHTp/+rxf2BjxNx5PjCdkxk3Jk8pOWU7BkZ3zCx3VWR3WoiV9vN+jBwAAwCChyAEAAAAgkNqiQ7OCQ9Dz6rWJSvF60d/gWv7ySOs2r4v0+ujM4ROH9dzoc8rOZyVJruUqkUzITJgyDH/JKtdy5RT8ZatKiyW/wFGRdEgSE3oAAACwhCIHAAAAgFXjz/+nP9fZr56VXbAVH4tLktyiq/h4XHv+mz166Dcf6vMIUXXk1SM6/shxZS9m5RQcxZNxxRIxyZS8sqeyU5ZbcJdncHj/P3v3HyPHed95/lM13T1D9pAccjhmd48yNGOJkkzxQo1J2bgcLWZtnIDkvMiR1NkU/xCO2hg4XO6UPWywwB4ERQdtDnd/bKDkEifZPXGBpSVLDHW4JIs4EAyLWUXrxGLknGQGkcKVuKa6R/ylmenu6uqq6qr7o9mjUbt/VFf/nnm/gIKaVc9Tz1PfrvlDz7ef5wkCPfHGE/qDP/mDYXcdAAAAI4QkBwAAAICR0mwprFZWrq7oD4/8oeRLM3tntGV2S3UTa9eXU3RUulnSD3/vh/rRv/2RvvnmN7Vtflufeo+wMkcyOv3d03r+6POyV21N7ZxSLBGTMWEo8IO12Ry1GRxPvPGEUosp6U+G3XMAAACMEpIcAAAAAEZGlATH8gfL+jdf/DdKziU1d2BO06lpxbfGFfjB2gbXpV0lTd2Y0uqHq/qDxT/QN9/8Zp+eAJ2Yf2heT5Wf0oVTF/TOS+8oNhVT4N/Zg8U05NmeDp46qOPfPj7kngIAAGBUkeQAAAAAEEotAdFun42w5ZrV69TvH/p9zSzMKHMko9337dZ0eloTiQl5JU+lj0uybliKT8Wrm1ob0upPVvX7h35f+p87bgp9cuLFEzrx4gktvbWk7Jt39lI5klHqUGrIPQMAAMCoI8kBAAAAoK1a0qKWuFifjFifnGhXrv5z7Xrt3+vL1F9rVO+P/8kfayI2obkDc5r7/JxSh1LasbBDFa+i0s2S4ktxGWZ1+aOKW5Fne/JsT8sfLEt/KumXehAc9EzqwZRSD5LYAAAAQHgkOQAAAACE0myWRf35sOV6ce1H//ZHmn9oXtPpaW2/a7t2fm6nZj47I/tju7pcVcmVU3TkFBwltiWUmE4okUxoy8wWuX/YQf+CAAAgAElEQVTjkuQYc42SYp2WC3OPRrOMwrYNAACA/jKH3QEAAAAAiOL6O9cVS8Q0tWNKienE2n4OTt5Rxa3IkCFzwpQZMzURn9BEYkITk3eOqQkZE4Z0Y9hPgaiazQbqpFwteVE7Gt2j2bl29QAAADAYzOQAAAAA0NLP/MzP6NatW2v/np2dlST97d/+rX7u535uaJ9zl3KSqWqyIpDckivrhiXf9eX7vpyCI8/xFFSCTw1wG2Y1+WEYhowsg9PjrH6ZtE7LtZuBEXV/mUZyl3LKXrqz38jhjNKL6Ybl9j4X7f5Xn2xf9+qT7e/T6B5h6nVbN8q9O7l/1L7V1xt0e/1qs9t4trpfu3t0GtNu3+tBfRe9fkf7HRcA2EiYyQEAAACgqV/7tV/T0aNHlc/n146a999/f6ifHcuRIUO+58spOip9XFJhqaDV7KqKHxVV+rgkZ9WRa7mqlCvV5EfFV+Df+fW9DAUOywyhsUZLVEVx/uR5PWM8o7NfPqs//1/+XH/+T/9cZ4+e1TPGM/qj/+6PetDT8NoNija7HqZe1LphtLpHq7bb1e/2vp3WG5X49/q5w5aPGtNuDOq76Camw4gLAGw0zOQAAAAA0NSOHTu0Y8eOhtd++Zd/eaif04fSCoJAbtGVvWzLum7JMAwl8gmZMVMVtyK36Kq8WpZTcOSWXFXsinzHl+/5qgQVGRlmcqBqfVKjXYIjzOyOa29c09mHzyq5O6mFowtKTCdkmIZ8tzrLyF62deXVK3p28lmdef2MMkcyktr/8j7Mr7MblanV3/tc6+v19dvVC9tut9rFpV/PVbvW6XPU33PU4h81nr3Qq5i2EzWmUet1G9NBxQUANiJmcgAAAAAYS5nDGbl2NcFRulVS4aOC8tn82lH8qCjrpqXSx6VqoiPvyLEcuXY12SFXCjLM5EDzjcVrR+3f663fk6PetTeu6YWvvaD0Ylr7vrpP8w/NK3M4o9ShlObun9Oue3ZpZt+MZvfPKrknqXOPnNOHf/1h/x5Q4QdC68uFXTKnWbleDMCGGRju9B5hEgZRB9U7GQBvV64f8e9FPMO2X3/vYQzIdxrTKPW6iemw4gIAGwlJDgAAAABjyYybuvu/vluFXEHF60UVl4oqZKuJjkKuoMJS9XzpVulTiY7a7A7tl8REjrHWLAHRSblGCY71CYz1+3k0u3f9tbMPn9Wuu3cpvZhW5nBGd33pLqW/kNbc5+e08+6dmtk7o+3z2zWdmta29DbFt8T1/NHnwz10H4QdKB+3JXVG9bnq2xvVfoY1igP0UWM67t8FAGxWLFcFAAAAYGx9/f/5un4z+ZsqXi9KkjzbU3w6ron4hGRIvufLs71qYiNfVnmlXE12WI70jSF3Hl2pLRlVS1ysTzSsTzy0K7f+v/XXm2lV/vzJ80rOJTW7f1a779ut1GJKM3tnVHEqKl4vKjYVk2FU95KpOJXq+2m5sldtvfKNV3T8O8c7DcXQdPIrfvTesOM/7OQQ710VcQEAkhwAAAAAxlhsKqZHv/Oo/vhX/lhBJZBXupPkSExUB7orgTzHk1fy5BQclfNlebanr7/ydb30ty9JrFY11polJBrNzOikftT7SdLlC5e1cHRByT1JbZvfppm9M9qxsEP2sq2KU5FruXLyjhLTieqRTCiejGtq55TefvntviU5BjEgPYzBVn5R/4lexL+TTcSjttEro9CHMIaVDBr1uABAL5HkAAAAADDW7n/0fhkThs5/47zKq2VN7pjUxOSEDNOQfKniVX8tX14tyy25+vqFr+vef3yv9LfD7jk2mtylnOJb40pMJxTfGpcZN1VxKrKXbXm2p8C/M6tkwpAZM2XGTZkJU7FErHpMxbT01pJSD6a66kerQdV+DHw2a2+Qg62jMKB79cnqMzfaYLqfA939iH/UDe77ZZDx7Jd+xGsjxAUAeoE9OQAAAACMvfuO36d/9tE/0+y9syosFbT8wbKW31/Wx1c/1upPVlW6XdJnDn5G//zWP68mODDW7r33XuVyubWjZv252vn33ntv7frFixf7+vm1F1+TDMkwDQV+ILfoqni9qHw2L+umJafgyLM9+Z6vwA+k4M7SV6ZkTFTrZN/MRg3L0NU2UB7URsqj/Iv1WrKjdgxCt/EPG89BD6S32zx9lAf2+/mOjnNcAKDXmMkBAAAAYEPYsnOLnviPT2j12qo+/KsPdfUvrkqS9j68V/MPzWv7XduH3EP0wtNPP61sNttwyah25/r9uXpC8l1fbsFV6XZJsamYXMuVYRifzCgquvJsTxW3Uk14VIJq0qNHmg2oNptl0K82W81s6NaoJjiabUzd7w2ru41/pwmOUYh77flG1bBiNepxAYB+IMkBAAAAYEPZftd2bb9ru+4/cf+wu4I+yWQyoc/v379/7fOxY8f6+/mxY3r/W+/LKTgqLZc0eWNShmHIyTsyJqqbjbtFV+XVspyiI9f6dLLDMA1ljjR+tl7oZ9JhkEZpoL2ZUe5bvSjxbDWIPg7fT78RAwAYLJIcAAAAAAD0QHoxLddyZS/bsm5aik3G5Hu+EtMJmTFTgR/Isz05RUfllbKcwp1Eh+XJs6tH6lB3+3FsdOM8eDyKfR/FPo07YgoAg8eeHAAAAAAA9MiBRw9U9+C4YamwVFA+m68eubwKSwUVbxRl3bRU+rik8kp5bVaHvWLr4KmDQ+lzu6WUmg3aDnoQt9PB46jPNWjDin+U56/f96PRHiCD3pOlWT9blWkX06jvzCi8UyxVBWAzIskBAAAAAECPnHz5pFzLXUtq5LN55T+sJjoKSwUVPyrKumGpdKuk0u1qosNesSVfOv7t433tW5jBz/oyYQdMG5Xr5YBvt/eK+lzdtlN/vtNNvfsV/1EYjA+jXTylwcW0Xb1BxrQXcQGAjYTlqgAAAAAA6KEzr5/RuUfOKX8tL9dyFU/GFUvEJFMKKoEqbkWe5a3N4AiCQE+88UTP2m83GNtqk+pm9ZsNmLar1wvr7xvm1/v156I8V5hB5Gb1o8RhkPHvJp7diBrTqO9Vv2La7jvvNKaDjgsAbETM5AAAAAAAoIcyRzI6/d3TKtwsaPmD5bUlqwq5wtpsjuKNovK5vNyCq8dffVypxf7vxdFuGaFWg77t7hulvUGJ+ly9bqubOParzVHX7n0dRkxHQbdxAYCNhpkcAAAAAAD02PxD83qq/JQunLqgd156R7GpmAI/kCQZpiHP9nTw1MHQS1SFGbTsxcBm1Hv0c1B1GM/VTZvDqNvpvhr9EDVx0O19+1V/UDEdxb85ABg3JDkAAAAAAOiTEy+e0IkXT2jprSVl38xKqs70SB3q/8wNAACAzYAkBwAAAAAAfZZ6MKXUgyQ2AAAAeo0kBwAAAAAAGJhONkxmSR4AANAOG48DAAAAAAAAAICxxEwOAAAAAAAwMMzOAAAAvcRMDgAAAAAAAAAAMJZIcgAAAAAAAAAAgLFEkgMAAAAAAAAAAIwlkhwAAAAAAAAAAGAskeQAAAAAAAAAAABjiSQHAAAAAAAAAAAYSyQ5AAAAAAAAAADAWCLJAQAAAAAAAAAAxhJJDgAAAAAAAAAAMJZIcgAAAAAAAAAAgLFEkgMAAAAAAAAAAIwlkhwAAAAAAAAAAGAskeQAAAAAAAAAAABjiSQHAAAAAAAAAAAYSyQ5AAAAAAAAAADAWCLJAQAAAAAAAAAAxhJJDgAAAAAAAAAAMJZIcgAAAAAAAAAAgLEUG3YHAAAAAAD9YxjG2ucgCDout/58o3u0un/YtgEAAIComMkBAAAAABtULclQSzDUJyzClguCYO2or7f+2vp6ra4BAAAAvcJMDgAAAADYwGqJiXaJhrDlGtXp9FozuUs5ZS9lJUmZwxmlF9MNy+19ruNbS5KuPtm+7tUnO7vn+vu1q9uo7Sh1wtTrRLdtRHmuRvUG3V4/2+zl99bNOxb1/eqmfj++i16/o1H72EmbAIDBIckBAAAAAGip30tSnT95XpcvXFZ8a1wyJN25jWu5OvDoAZ18+WSk+0ax97nuBr47LduqvVb3r13rdsC12zZ6/VyttOtrr/sZtc1efm9hYxU1pt3odUz7Ec9+vWut2gQADB5JDgAAAABAU82WqGp0vf5au/PX3rimsw+fVXJ3UgtHF5SYTsgwDfmuL6fgyF62deXVK3p28lmdef2MMkcyktoPaIYZfGw1mNpJoiOMZr/ED9tev/vaLp5hBp+7ea5OB6Lr79nvfjZqs/6+Ycp32mYUvYppO1FjGrVet/HsZVz6/R0CADrHnhwAAAAAgL5pleB44WsvKL2Y1r6v7tP8Q/PKHM4odSilufvntOueXZrZN6PZ/bNK7knq3CPn9OFff9jXvnYyYBnl19z1ZcMMrDcr06vB1U6TK2HKdfNczbSKd7/62e47jvLd9OsdixLTXug0plHqdRPPqHEZxN8eAKB3SHIAAAAAwAZWW06q3T4bYcs1qtPqerMlrM4+fFa77t6l9GJamcMZ3fWlu5T+Qlpzn5/Tzrt3ambvjLbPb9d0alrb0tsU3xLX80efD923QehkXf92A6bDWG6oG6P6XPXtjWo/wxrFAfWoMR337wIAMLpYrgoAAAAANqjaJuK1ZESzpaXalau/Z/39G11rlDSpXT9/8rySc0nN7p/V7vt2K7WY0szeGVWciorXi4pNxWQYhnzPV8WpyLM9uZYre9XWK994Rce/c7y7wHSJQdiNb9j7Lgw7OTSKyRUAAJohyQEAAAAAG1izmRT158OWi3r/9S5fuKyFowtK7klq2/w2zeyd0Y6FHbKXbVWcilzLlZN3lJhOVI9kQvFkXFM7p/T2y2/3LckRZmB52IPfg+wHyZxP9CoJMC7v2Cj0IYxhbrg+6rEBgM2EJAcAAAAAYGByl3KKb40rMZ1QfGtcZtxUxanIXrbl2Z4C/86skglDZsyUGTdlJkzFErHqMRXT0ltLSj2Y6qofUTaNDnu9Hxr1d1D9GIXB3KtPVmPQaLPnfg50N7t3twPdo/SODTKe/TLoZN8o/E0AAD7BnhwAAAAAsAHlcrlPHb0+/+Mf/3jt88WLF0N/zl7KSoZkmIYCP5BbdFW8XlQ+m5d105JTcOTZnnzPV+AHUnBnyStTMiaqdbJvZnsUpc6M2uDvIAb3R3Ewt5bsqB2DUNuIupsNvsPEdNDvWLuN3EftnV9vmO/oKMcFADYjZnIAAAAAwAbzzDPP6J577tGxY8d+6lqny0uFOd/pZwWS7/pyC65Kt0uKTcXkWq4Mw5BneyqvluUWXXm2p4pbqSY8KkE16dEjzQZG280WGNagf6N2m/W1W8N+1maabUzd7w2rmyUCOol/JwmOUYh77flG1SBjNci/PQBANCQ5AAAAAGADevfdd/XYY4/91PlMJtOwfKfnH3jggbXP65Mp7T5nDlfv5xQclZZLmrwxKcMw5OQdGRPVzcbdoqvyallO0ZFrfTrZYZiGMkca96kX2g1etxr4HfQgdacD7WGM0kB7M6Pct0Y6jekovWOjaBRi0I+/PQBAdCQ5AAAAAAADk15My7Vc2cu2rJuWYpMx+Z6vxHRCZsxU4AfybE9O0VF5pSyncCfRYXny7OqROtTdfhxobBQGj6Ma1b6Par/GFfEEADRCkgMAAAAAMFAHHj2gK69eqSY2JqobjyemEzLjphRIFbci13LlFKqJjtqsDnvF1sFTB4fS56jLDIWdGTLsQdtO+zEuzxW1n71YrilKTHt1r261evZuYxr1nRmVdwoAMHrYeBwAAAAAMFAnXz4p13KVz+VVWCoon80r/2Fe+Wz138WPirJuWCrdKql0u6TySln2ii350vFvH+9r3waxmXfYtrq9Hla3g8edPldUze4btv9R+9mo3EYYkG8XT2lwMQ37ro9CkifMdQDAYDGTAwAAAAAwcGdeP6Nzj5xT/lperuUqnowrlohJphRUAlXcijzLW5vBEQSBnnjjiZ61326QspeDqetnBTRqN8wv+NvdP6r192/36/1G56I8V5jB9Wb1owwuR+1nu3rNdBPTqKLGNOpgfb9i2u477zSe/X7XRjmJBQCbCTM5AAAAAAADlzmS0envnlbhZkHLHyxXZ3Nk8yrkCmuzOYo3isrn8nILrh5/9XGlFvu/F8fVJ/szcNlq0LfTOuuvD3uQNcpz9aOtMLHq5Hwv2hx17d69YcR0VIzD3x4A4BPM5AAAAAAADMX8Q/N6qvyULpy6oHdeekexqZgCP5AkGaYhz/Z08NTB0EtUhRl0HOTgey/a7vdAai/u3+k9umlzGHUH+XxR7zmoZxtGu+P2vgAABo8kBwAAAABgqE68eEInXjyhpbeWlH0zK6k60yN1qP8zNwAAADDeSHIAAAAAAEZC6sGUUg+S2AAAAEB4JDkAAAAAbHqGYax9DoKg43Lt6re6HrZtIIxONpJmOR4AALARsPE4AAAAgE2tlmSoJRjWJx3ClguCoGmCwjCMtetBEHyqXqtrAAAAANpjJgcAAACATa+WoGiXaAhbrlGdTq81k7uUU/bSnX0rDmeUXkx3fA9sXMzOAAAAmw1JDgAAAAAYA+dPntflC5cV3xqXDEl38iOu5erAowd08uWTQ+0fAAAAMAwkOQAAAABgQGrLU3Vy7dob13T24bNK7k5q4eiCEtMJGaYh3/XlFBzZy7auvHpFz04+qzOvn1HmSKbfjwEAAACMDJIcAAAAADAAURMcL3ztBaUX05rdP6vknqTiW+MK/EBuwVVpuSTrpqXEdEL5XF7nHjmn09893e9HAQAAAEYGSQ4AAAAAm14tydBun42w5ZrV6/Ta2YfPKr2YVnoxrd337da2+W0y46bcoqvS7ZImb0wqNhmTOWFKkvLX8nr+6PPSv+ioewAAAMDYIskBAAAAYFOrJS1qiYv1CYf1CYh25eo/167X/r2+TJhr50+eV3Iuqdn9s9p9326lFlOa2TujilNR8XpRsamYDMOQ7/mqOBV5tifXcmWv2nLOOxJbdAAAAGATIMkBAAAAYNNrNpOi/nzYcr24dvnCZS0cXVByT1Lb5rdpZu+MdizskL1sq+JU5FqunLyjxHSieiQTiifjmto5Jeey0/S+AAAAwEZiDrsDAAAAAIBPy13KKb41rsR0QvGtcZlxUxWnInvZlmd7Cvw7s0omDJkxU2bclJkwFUvEFEvEpJhkLHW2pBYAAAAwjkhyAAAAANi07r33XuVyubWj5uLFi0P9nL2UlQzJMI3qJuNFV8XrReWzeVk3LTkFR57tyfd8BX4gBXeWvDJVTXwYppTtPB4AAADAuGG5KgAAAACb0tNPP61sNttwyaj154b1WYHku77cQnWT8dhUTK7lyjAMeban8mpZbtGVZ3uquJVqwqMSVJMeAAAAwCZBkgMAAADAppXJZBqeP3bs2FA/Zw5X++UUHJWWS5q8MSnDMOTkHRkT1c3G3aKr8mpZTtGRa9UlO4JAQYZkBwAAADY+khwAAAAAMGLSi2m5lit72ZZ101JsMibf85WYTsiMmQr8QJ7tySk6Kq+U5RTuJDosr7pnhxdIqWE/BQAAANB/JDkAAAAAYAQdePSArrx6pZrYmKhuPJ6YTsiMm1IgVdyKXMuVU6gmOmqzOuwVW3pg2L0HAAAABoMkBwAAAACMoJMvn9Szk88qn8tLkjzbUyKZkJkwZRjVJas825NrVZetKq+UqwkOX9IJSaxWBQAAgE3AHHYHAAAAAACNnXn9jNyCq/y1vPLZvFazqypkC8rn8iosFVS8UZR1w1LpdknWbUtBEOiJN54YdrcBAACAgSHJAQAAAAAjKnMko9PfPa3CzYKWP1hWPltNdhRyhWqS46OiijeKyufycguuHn/1caUW2YwDAAAAmwfLVQEAAADACJt/aF5PlZ/ShVMX9M5L7yg2FVPgV9eiMkxDnu3p4KmDOv7t40PuKQAAADB4JDkAAAAAYAycePGETrx4QktvLSn7ZlZSdaZH6hAzNwAAALB5keQAAAAAgDGSejCl1IMkNgAAAACJJAcAAAAAbGiGYax9DoIgUrkw9zAMI1I9AAAAoBtsPA4AAAAAG1QtyVBLMKxPOoQtV0te1I5G92h2rl09AAAAoFvM5AAAAACADayWuGiXaGhWrt0MjFoyoxdJjNylnLKX7uw3cjij9GK6Ybm9z0W7/9Un29e9+mTza1HrNqvXqq0oum2nUf0wdevrDbq9frXZ6+9t/f3a3aPTmHbzXjer34/votfvaD/jMqi/WwBA90hyAAAAAAAiabREVRTnT57X5QuXFd8alwxJd27pWq4OPHpAJ18+2XUbYe19rreDmK0GWWvXetFet+00q98qHlGTTe362ut+Rmmz199b2FhFjWk3eh3TZvW6iemg4zKov1sAQG+Q5AAAAAAAhLI+qdEuwRFmdse1N67p7MNnldyd1MLRBSWmEzJMQ77ryyk4spdtXXn1ip6dfFZnXj+jzJGMpPYDoWEGH1sNwrYb3I0yuNlNe920EaadZjMMOo1HpwPRzRIL/epnozbr79uubKftRdWrmLYTNaZR63Ub00HFpVWdQXz/AIDOsCcHAAAAAKCtZhuL147av9dbvydHvWtvXNMLX3tB6cW09n11n+YfmlfmcEapQynN3T+nXffs0sy+Gc3un1VyT1LnHjmnD//6w/49oPozYHn1yeb37WV7vUjK1JcLkzCIOqjeyQB4u3Jhlyzq5HvoZZKr0+TbMAbOO41plHrdxHTQcel1khMA0F8kOQAAAABgA2uWgOikXKMEx/oExvr9PJrdu/7a2YfPatfdu5ReTCtzOKO7vnSX0l9Ia+7zc9p5907N7J3R9vntmk5Na1t6m+Jb4nr+6PPhHhqhhU0ADHu5oFHtZ1ijODAeNabj/l0AADYelqsCAAAAgA2qtmRULXGxPtGwPvHQrtz6/9Zfb6ZV+fMnzys5l9Ts/lntvm+3UospzeydUcWpqHi9qNhUTIZhyPd8VZyKPNuTa7myV2298o1XdPw7xzsNRU9F3Wgbgzfs/ROGnRzi3awiLgCwsZHkAAAAAIANrFlCotHMjE7qR72fJF2+cFkLRxeU3JPUtvltmtk7ox0LO2Qv26o4FbmWKyfvKDGdqB7JhOLJuKZ2Tuntl9/uW5Kj2420+7mMUTf4Rf0nejHY3ekm4sMcUB+FPoQxrGRQP79/AMDgkOQAAAAAAAxM7lJO8a1xJaYTim+Ny4ybqjgV2cu2PNtT4N+ZVTJhyIyZMuOmzISpWCJWPaZiWnprSakHU131o9VAZSfr8Xcy4Nmo7KjsMzDIPux9rvGmzf0cPG52724Gu6NucN8vg4xnv/RrX5z1ehWXUfh7AgBUsScHAAAAAGxA9957r3K53NpRs/5c7fzf//3fr12/ePFiXz+/9uJrkiEZpqHAD+QWXRWvF5XP5mXdtOQUHHm2J9/zFfiBFNxZ+sqUjIlqneyb2ahh6Uq7DaqjDJ72eyB6lH/JX0t21I5BqG1gHXUj67DxHHSCoR/v5qD08x3tdVxG+e8JADYzZnIAAAAAwAbz9NNPK5vNNlwyqt25fn+unpB815dbcFW6XVJsKibXcmUYhjzbU3m1LLfoyrM9VdxKNeFRCapJjx5pNkjZbJZBu3uFGSxtdM8o7YU1qgOyzTam7veG1c0GvMN+B50mOEYh7mHfzWEZVqyixGWUvlcAwKeR5AAAAACADSiTyYQ+f9999619PnbsWH8/P3ZM73/rfTkFR6XlkiZvTMowDDl5R8ZEdbNxt+iqvFqWU3TkWp9OdhimocyRxs/WC50Meo9ye+MwIDvKfasXJZ6tBtHH4fvpt3GKwTj1FQA2I5IcAAAAAICBSS+m5Vqu7GVb1k1LscmYfM9XYjohM2Yq8AN5tien6Ki8UpZTuJPosDx5dvVIHepuP46NbpwHZEex76PYp3E3TjEdp74CwGbFnhwAAAAAgIE68OiB6h4cNywVlgrKZ/PVI5dXYamg4o2irJuWSh+XVF4pr83qsFdsHTx1cNjd/ymjtBxQpwOy7ZaIGpUB3qj97LbfUZ6/ft+PRnuARN0TpFOt3s1uYxr1nRmFdyrs3+wo9BUA0B5JDgAAAADAQJ18+aRcy11LauSzeeU/rCY6CksFFT8qyrphqXSrpNLtaqLDXrElXzr+7eN97Vu7gdtW55vtuRGlvSi6HZCt70u/kjfdJlSi9rNRuVZtjssAd9R3s9U9osY07Ps+zARP2LiMy/cPAGC5KgAAAADAEJx5/YzOPXJO+Wt5uZareDKuWCImmVJQCVRxK/Isb20GRxAEeuKNJ3rWfrvB2CgJi6htNWsvahthfr1ff65Wp1Hddr/Ib3e+V7GM2s929RrpJp7diBrTqO9mv2La7jvvNKaDjsuwvn8AQDTM5AAAAAAADFzmSEanv3tahZsFLX+wvLZkVSFXWJvNUbxRVD6Xl1tw9firjyu12P+9OJotI9RqMLPV0kPtBkEHtWxRO1H73+u2wsSrk/O9aHPURX0329XvV71B6TYuAIDxwUwOAAAAAMBQzD80r6fKT+nCqQt656V3FJuKKfADSZJhGvJsTwdPHQy9RFWYQctuBjaj1h3EYGov2uj0HsOIZTd1O91Xox+iJg66vW+/6g8qpoOOCwkQABgvJDkAAAAAAEN14sUTOvHiCS29taTsm1lJ1ZkeqUP9n7kBAACA8UaSAwAAAAAwElIPppR6kMQGAAAAwiPJAQAAAADAiOlkw2SW1gEAAJsZG48DAAAAAAAAAICxxEwOAAAAAABGDLMzAAAAwmEmBwAAAAAAAAAAGEskOQAAAAAAAAAAwFgiyQEAAAAAAAAAAMYSSQ4AAAAAAAAAADCWSHIAAAAAAAAAAICxRJIDAAAAAAAAAACMJZIcAAAAAAAAAABgLJHkAAAAAAAAAAAAY4kkBwAAAAAAAAAAGEskOQAAAAAAAAAAwFgiyQEAAAAAAAAAAMYSSQ4AAAAAAAAAAHPSBWIAACAASURBVDCWSHIAAAAAAAAAAICxRJIDAAAAAAAAAACMJZIcAAAAAAAAAABgLJHkAAAAAAAAAAAAY4kkBwAAAAAAAAAAGEskOQAAAAAAAAAAwFiKDbsDAAAAAIDhMAxj7XMQBB2Xa1U/zL3Dtg8AAAA0w0wOAAAAANiEagmGWnJhfcIhbLkgCJomJ1pdq92nVoYEBwAAAKJiJgcAAAAAbFK15EIQBE2THJ2UC6uW4OhE7lJO2UtZSVLmcEbpxXTDcnufi9anq0+2r3v1yebXmtWNUqeTe4QVpX/t6oepW19v0O31q81u49nqfu3u0WlMu3mvm9Xvx3fR63c0alw6/Q67eecAAL1BkgMAAAAAMHBhl6o6f/K8Ll+4rPjWuGRIulPUtVwdePSATr58ss89/cTe5xoPXrYaRK5dG+agZ7f9a1a/WTzatdlKu772up9R2uz19x02VlFj2o1ex7Qff0NR4tKL77CbdxUA0FskOQAAAAAAA1e/t0d9ouPaG9d09uGzSu5OauHoghLTCRmmId/15RQc2cu2rrx6Rc9OPqszr59R5khGUvuB0DADj60GYVsNXnZaL8wgca8GStvFJczg8/rrYeKxvk6nA9HNEgv96mejNuvv265sp+1F1auYthM1plHrdRvTKHGJ+rce9h4AgMFgTw4AAAAAwEi59sY1vfC1F5ReTGvfV/dp/qF5ZQ5nlDqU0tz9c9p1zy7N7JvR7P5ZJfckde6Rc/rwrz/sa5/aDa52kvgYtDADw53eI0zCIOqgeicD4O3Khf01fiffXy/iGbb9+nsP433qNKZR6nUT0yhx6fZvNsp7AwDoH5IcAAAAALBJ1ZaMarfPRthyvXL24bPadfcupRfTyhzO6K4v3aX0F9Ka+/ycdt69UzN7Z7R9frumU9Palt6m+Ja4nj/6/ED6Ngij8ivwsAO5g+5vfXuj2s+wRnFAPGpMx/27AACMJ5arAgAAAIBNqLaJeC1x0Wz5qHbl6j/Xrre6Vr+B+fp7nj95Xsm5pGb3z2r3fbuVWkxpZu+MKk5FxetFxaZiMgxDvuer4lTk2Z5cy5W9auuVb7yi49853oPojIZRHPzeSIa9X8qwk0O8X9EM+70BAPw0khwAAAAAsEk12/C7/nzYcmGvtbp++cJlLRxdUHJPUtvmt2lm74x2LOyQvWyr4lTkWq6cvKPEdKJ6JBOKJ+Oa2jmlt19+u29Jjm430h7VAVF+Uf+JXiQBOt1EfBQ2pB/Vd7NmWMmgsHEheQQAw0eSAwAAAAAwEnKXcopvjSsxnVB8a1xm3FTFqchetuXZngL/zqySCUNmzJQZN2UmTMUSseoxFdPSW0tKPZjqqh+tBlXD7vHQSZ1G9Qc9UDoKA7NXn6w+f6MNn/s50N3s3t18F1E3rO6XQcazX/oVr6h/s/14bwAA0bAnBwAAAAautvRNmH0AGpVrVX/9tTBlgM0ol8t96pCkH//4x2vXL168OJTPr734mmRIhmko8AO5RVfF60Xls3lZNy05BUee7cn3fAV+IAV3lsIyJWOiWif7ZrbjePTbqA4oj/JgbC3ZUTsGobYZddQNvsPGc9DvQ7vN00f1/ZSG9452EpNu3xsAQPeYyQEAAICBWr82fy3R0GjZmlblGq35X1N/r0YJknbL6AAb2TPPPKN77rlHx44d+9T59X8Xw/pcPSH5ri+34Kp0u6TYVEyu5cowDHm2p/JqWW7RlWd7qriVasKjElSTHj3SbKCy2SyDdnXD1Bu0UU1wNNuYut8bVjdLBIT97jpNcIxC3GvPN6oGFatu/ma7fW8AAL1BkgMAAAAD12zz4ajlmqlPaERNcOT+pvpL9/RiuuO6wCh699139dhjj33q3AMPPLD2eX0CZKCfHzum97/1vpyCo9JySZM3JmUYhpy8I2Oiutm4W3RVXi3LKTpyrU8nOwzTUOZIpt3jRxZ18DJsvUEN6o7SQHszo9y3elHi2Sq5MA7fT78NOwYkKgBgvJDkAAAAwKayPlnSKuHxg3/1A/3gt3+glasrmtw2KUkq58ua2TujL/7TL+pLT36p730FNpv0Ylqu5cpetmXdtBSbjMn3fCWmEzJjpgI/kGd7coqOyitlOYU7iQ7Lk2dXj9Sh7vbj2OiGPXjcjVHs+yj2adwRUwBAp9iTAwAAABtSs1kbQRCsHY1mhzgFR7+z/3f0H/7lf1BsKqa9X96r9OG09vzcHqUeTKniVfTa06/pd+//XbmWO4hHATaVA48eqO7BccNSYamgfDZfPXJ5FZYKKt4oyrppqfRxSeWV8tqsDnvF1sFTB4fd/cgGsWxQp4PH7ZaIGpXB6Kj97LbfUZ6/fv+GRns5DGpvh1bvXLcxjfrOjMo71coo9w0ANiuSHAAAAMAd9se2fufu35Eq0sKXF7TvF/Ypczij1KGU5g7Maff+3Zr7/Jx27tup4vWifvtzvy17xR52t4EN5eTLJ+Va7lpSI5/NK/9hNdFRWCqo+FFR1g1LpVsllW5XEx32ii350vFvH+9r39oN3HZar5F+DaB2O3hc/wz9Ssp0m1CJ2s9G5Vq1OQ6D8VK4d3ZQMQ37dzLsBE+Y663Kjcu7AQAbCctVAQAAYOBqsyza7bMRtlyv/O6B39XkzKTu+uJdmr13Vtsy2zSRmJBne7I/vrN8ztaYJmITkiGt/OcVfeuBb0n/ZCDdAzaNM6+f0blHzil/LS/XchVPxhVLxCRTCiqBKm5FnuWtzeAIgkBPvPFEz9pvN8DZatC703qDsL5vYX69X3+uVqdR3Xa/yG93PmosG/UjSj/b1Wukm3h2I2pMoyak+hXTdt95pzHtZ1x6+d4AAPqHmRwAAAAYqNoSUrXERf3G4GHL1c6v/7z+erOlqmrl68u8+uuvqmJXtOe/2KO5A3NKfyGt+S/Oa8/BPdr5uZ3aftd2TaemlfxMUlt2b9GWXVs0/ZlplW6XpO91FRIAdTJHMjr93dMq3Cxo+YPltSWrCrnC2myO4o2i8rm83IKrx199XKnF/u/F0WwZoXaD2e2WHxr1X363GugdZFth4tzJ+V60Oepa9X9YMR0F3f7NtrrHRnhvAGDcMJMDAAAAA9dsw+/682HLdXK9YfKjEujSv76k3ffu1vbMdu1Y2KHZ/bPauW9nNYlhSJVyRa7lyik4crY5cqarx+TMpNwfujJ+YTCzTYDNYv6heT1VfkoXTl3QOy+9o9hUTIF/J/lpGvJsTwdPHQy9RFWYQcduBiaHVXdQ9+/0HuMWy0731eiHqIPq3d63X/UHFdNhxaVX9wAAdI8kBwAAADa9W+/dku/4mto5pcS2hOJb4jJkyC26CiqBDNOQGTNlxk1NJCaqx2T1iE/FZVQM+bf9YT8GsCGdePGETrx4QktvLSn7ZlZSdaZH6lD/Z24AAABg9LFcFQAAAAZmz549WllZWTtqav/+h3/4h7Vz77333sA+v/Xv35IMaSI+IUnybE/WTUv5bF7WbUtO0VGlXFHgB9WZIEb1l+TGRDX5YRiGlO08HgDCSz2Y0uKvLGrxVxZJcAAAAGANMzkAAAAwEN/85jd169Yt3bhxY+3cjh07JGnt3I9+9CPdfffdkqQf/vCHuueeewby+R/e/gcZpqGKW12SqvRxSfGluFzLlQzJK3lyCo5cy1WlXJHv+vIrvuRXl78yZcq3mckBYDA62eiY5XQAAMBGR5IDAAAAA5FOp5VOpxteqyU2av+VpMcee2xgn3/piV/SufPn5Fqu7BVb1g1LhmnIKTgyJgz5ri+n6Ki8WpZTdOSVPFXKFVXcinzPVyWoSPywHAAAAAAGjiQHAAAANr3M4Yy8sqfySlmlWyXFpmLyfV9O3pEZM+VXfHm2Jyf/SaLDtVx5djXZEXiBgkzrzdABoFeYnQEAAPAJkhwAAADY9GJbYtr75b26/uPrSmxLyJww5bu+ysmyJuITCoJAvuPLLbkqr5Zlr9hyCk51dke+LO2TNDHspwAAAACAzYckBwAAACDp5Esn9Vvzv6XiR0VJklf2FE/GNZGoZi98rzqbwy2uS3SsOvJsT3p0mD0HAAAAgM3LHHYHAAAAgFGQnEvqkd96RIWPCirkCspn82tHIVdQ8aOirOuWrJuWSrdLsj+25RQd/eL/9YvSlmH3HgAAAAA2J2ZyAAAAAHcc+R+PaCI+oT978s9UXi1rcvukJiYnZJiG5EsVr7K2N4dne/rF3/tFPfjfP6g/feZPh911AAAAANiUSHIAAAAA6yx+c1E/c/Rn9NIvv6R8Nq+KU/lkXw7P10RiQtsz23XqT05p1/5dw+4uAAAAAGxqJDkAAACAOnP3z+lX//5XlX0zq5+8/hNdff2qJGnvl/dq4ecXlP5Cesg9BAAAAABIJDkAAACApjKHM8oczuiLv/bFYXcFAAAAANAASQ4AAAAAaMEwjLXPQRBEKhf12voyrdoGAAAANitz2B0AAAAAgFFVS0DUEgzrExJhy7W7FgTB2tHo/s3aBAAAAECSAwAAAABaqiUn2s2kaFUu7D3qMYMDAAAAaI3lqgAAAABgSOpnb6xPaHSS4Pj4ysf64e/+UO9//33dvnJbkrTr7l3a9wv79ND/9JBmPjvT244DAAAAI4IkBwAAAAAMSX0io/bvThIcr/76q/qb//tvpECampnSzs/ulF/xZX1k6dIfXtJbz7+lw//DYX3lN7/Sr8cAAAAAhoYkBwAAAACMoPq9OBolPv7dV/+dcn+TU+pQStN7ppVIJiRJru2qvFqWfduWddPSX/3OX+mjtz7SY3/22MD6DwAAAAwCSQ4AAAAAaGH97Iqo5cLeo6Y+mdEowXHukXNa+v+WtPfYXs3eM6vp9LRiUzFVnIrKK2VZtyxZ2yxNTE3IiBm6+vpVvfDfvCAdCdUFAAAAYCyQ5AAAAACAJmqJiVpyotmeGa3Khbm2/t9h/N2Fv9O1/3hNC19e0Gce+Iz2HNyj7XdtlwzJXrZVvF5UbComwzQU+IF811fFqeiD1z6QtkvaHykcAAAAwMghyQEAAAAALTRLPNSfb5WgiHqtWZnvP/V97dy3UzsWdmjX53Zp7oE5ze6fVelWSflsXoEfqOJU5JZcuZYrp+jIKTgqJ8tyv+eS5AAAAMCGYQ67AwAAAACA8Aq5gm7/p9va+pmt2rJziyZnJtf24jBjpiYmJxSbiv30sSWmeDIu3ZaMYrhlswAAAIBRx0wOAAAAABgj2UvZtWSGMWHI93zZy7ZkSL7ny7M9+Z4vBdUltQzTkDlhyoxVD8M0FGTDLYsFAAAAjDpmcgAAAABAnYsXL+rSpUtrx+XLl/XMM8+sXR/m5+X/tCxzwqwuSVWubjJeWCoon83Lum6pvFyWW3Tl2Z4qbkVBJVDgB2tJjwljQrodKSwbRm2PlDCbyTcrF/VaJ+0DAACgPWZyAAAAAECd1157TUEQ6Dd+4zfWzj399NMj8fndP323OmOj5Km8WpZ105I5YcopODInTFWcipxCdQ8O1/ok2eF7vnzPV8WvSLs6CseGsn4D+FqiodG+KK3K1dcJe63RvwEAANAdkhwAAAAAMEYyX8jIr/gqr5ZVul1SfGtcgR8okU/IjJkKKoHckisn71SPoiPP8qrJDqeiwA8UZDb3IHstyVBLYHRaLupG8lESHLlLOWUvZSVJmcMZpRfTDcvtfa6j2665+mT7ulefbH4tat1m9Vq1FUW37TSqH6Zufb1Bt9evNnv9va2/X7t7dBrTbt7rZvX78V30+h3td1yitAkA/UaSAwAAAADGyHR6WrOfm1XpZknFHUWZMVMVt6LEdEIT8YnqMlZORU7RkbN657gzq8MtudJuSclhP8XmFTZZcv7keV2+cFnxrXHJkHSnqGu5OvDoAZ18+WSfe/qJvc/1dhCz1SBr7Vov2uu2nWb1W8UjarKpXV973c8obfb6ewsbq6gx7UavY9qsXjcxHaW4AMCwkeQAAAAAgDHzj/73f6QLj11QfFtchmHIK3trSQ5J8l1fru3KKTgqr5Zlr9pyCo68kif9t0Pu/AbTanZGo2vtlq669sY1nX34rJK7k1o4uqDEdEKGach3fTkFR/ayrSuvXtGzk8/qzOtnlDmSkdR+IDTM4HOrQdh2g7udJiXa9bdXiZWo7TSbYdBpPDodFG6WWOhXPxu1WX/fdmU7bS+qXsW0nagxjVqv25gOKi69aBMA+oWNxwEAAABgzNz7j+/VZx/+rFauriifyyufzSv/4Z3/5vIqfFSQdcNS6VZJ9se2ystlWbcs/exXf1a6Z9i93zg6TXC0c+2Na3rhay8ovZjWvq/u0/xD88oczih1KKW5++e0655dmtk3o9n9s0ruSercI+f04V9/2ItHaaofA9a9TJb0u536cmESBlEH1TsZAG9XLuySRc3KddqXXj5zo3sPYzmkTmMapV43MR1GXIb1XQBAOyQ5AAAAAGAMPfbvH9PCf7Wglf+8okK2UE1wZPMq5AoqfFRQ8aOiijeKsm5aKt4s6nNf+Zy+8f9+Y9jdHgm1JaNa7cfRrlyvExySdPbhs9p19y6lF9PKHM7ori/dpfQX0pr7/Jx23r1TM3tntH1+u6ZT09qW3qb4lrieP/p8x+2gtbAJgEH/ir2+vVHtZ1ijOFgeNabj/l0AwLhjuSoAAAAAGFOn/+y0vve/fk9v/t6bKnxU0MTkhGKT1f/N82xPlXJFE1sm9PO//vM69syx4XZ2RNQ2Ea8lLpotH9Wu3Pr/rr/e6lqrDczPnzyv5FxSs/tntfu+3UotpjSzd0YVp6Li9aJiUzEZhiHf81VxKvJsT67lyl619co3XtHx7xzvQXSiYyPi8dHLfU+6aX9Y7fFuVhEXABsJSQ4AAAAAGGNf+Zdf0eFvHtYPf++Hev977+vWe7ckSbv279LPfuVn9dCvPqTtd20fci9HS7NZFq32zwhzvt21VtcvX7ishaMLSu5Jatv8Ns3sndGOhR2yl21VnIpcy5WTd5SYTlSPZELxZFxTO6f09stv9y3J0e1G2t3sh9BP/KL+E70Y7O50E/FhDqiPQh/CGFYyaNTjAgCNkOQAAAAAgDG3Y+8OffX/+Oqwu4GIcpdyim+NKzGdUHxrXGbcVMWpyF625dmeAv/OrJIJQ2bMlBk3ZSZMxRKx6jEV09JbS0o9mOqqH60GVTvZO6AXg7ODGmgdhQHdq09WY9Zog+l+DnQ3u3c3g91RN7jvl0HGs18GsS/OOMYFANZjTw4AAAAAwKaQy+U+dTQ7/3d/93dr1y5evNj3z9lLWcmQDNNQ4Adyi66K14vKZ/OyblpyCo4825Pv+Qr8QAruLIdlSsZEtU72zWw3oYms3QbVnQyeDuqX5KP8i/VasqN2DEJtM+mom0qHjeegB9J7+W4OWj/f0XGOCwA0Q5IDAAAAALDhPfPMM/r+97+vIAjWjpr15xpdG8jnQPJdX27BVel2SYWl6mbyxY+KKt0qqbxallt0q3utuJVqwqMSVJMePVI/2L1+0LvTgc+oy1Rt1gRHswRD1MRDJ+02OxfmO+80wTEKcR+FPrQyrFiNelwAoBWWqwIAAAAAbArvvvuuHnvssZ86n8lkmv772LFjff+c216dVeIUHJWWS5q8MSnDMOTkHRkT1c3G3aKr8mpZTtGRa3062WGYhjJHPv0MvdRqOaVe2OwJjvVGuW/1osSzVeJkHL6ffiMGABANSQ4AAAAAAIYovZiWa7myl21ZNy3FJmPyPV+J6YTMmKnAD+TZnpyio/JKWU7hTqLD8uTZ1SN1qLv9OIaFBEd7o9j3UezTuCOmABAdy1UBAAAAADBkBx49UN2D44a1tlRVPptXPpdXYamg4o2irJuWSh+XVF4pr83qsFdsHTx1cNjd/ym9XOqoV30J20675ZpGZTA6aj+77XeU52+2FNr6e/R7aa6aVu9mtzGN+s6MwjvFXhwAxhlJDgAAAAAAhuzkyyflWu5aUiOfzSv/YTXRUVgqqPhRUdYNS6VbJZVuVxMd9oot+dLxbx/va9/aDdy2Oj/sQd1u26l/xn4NBHebUInaz0blWrU5CoPxYXTzbja7R9SYtqs3yJj2Ii4AMIpYrgoAAAAAgBFw5vUzOvfIOeWv5eVaruLJuGKJmGRKQSVQxa3Is7y1GRxBEOiJN57oWfvtBmNbDXpHbSfMr+qj6qad2j4kzeq2S960O9+rWEbtZ7t6jQzqe2vVbqvz9e1GTUj1K6btvvNOYzrouHTTJgD0GzM5AAAAAAAYAZkjGZ3+7mkVbha0/MHy2pJVhVxhbTZH8UZR+VxebsHV468+rtRi//fiaLaMUKuBzEEtPdRPrQazB9lWu/ai9rObNkddt+9mP2I6Cjb63yyAzYuZHAAAAAAAjIj5h+b1VPkpXTh1Qe+89I5iUzEFfiBJMkxDnu3p4KmDoZeoCjNo2c3AZpS6gxpI7UU7nd5j0LHstm6n+2r0Q9TEQbf37Vf9QcV0GHEhCQJgVJHkAAAAAABgxJx48YROvHhCS28tKftmVlJ1pkfqUP9nbgAAAIwTkhwAAAAAAIyo1IMppR4ksQEAANAMSQ4AAAAA2KQMw1j7HARBpHLNrq0/X3+9/lrYe2Jz62TDZJbVAQBg82DjcQAAAADYhGqJhGaJhzDlWl0LguBTR71m1wzD+NS1Zv0CAAAAJGZyAAAAAMCmVUswtEsmtCoX5h61xEW/5C7llL10Z9+KwxmlF9N9awvDw+wMAADQCEkOAAAAAMDA9WJJqvMnz+vyhcuKb41LhqQ7t3EtVwcePaCTL5/sQU8BAAAwykhyAAAAAAD6ptEsjmZLVNWutVui6tob13T24bNK7k5q4eiCEtMJGaYh3/XlFBzZy7auvHpFz04+qzOvn1HmSKa3DwUAAICRQZIDAAAAADBSWm1gfu2Na3rhay8ovZjW7P5ZJfckFd8aV+AHcguuSsslWTctJaYTyufyOvfIOZ3+7ulBPwIAAAAGhCQHAAAAAGxStRkU7WZOtCoX9h7d9G+9sw+fVXoxrfRiWrvv261t89tkxk25RVel2yVN3phUbDImc8KUJOWv5fX80eelf9Hz7gEAAGAEkOQAAAAAgE2olpioJSfqZ0/ULx/VqFyra/X3qT9f35dG1+rrnj95Xsm5pGb3z2r3fbuVWkxpZu+MKk5FxetFxaZiMgxDvuer4lTk2Z5cy5W9ass570hs0QEAALDhkOQAAAAAgE2q2Ybf7fbQ6PZa1PtdvnBZC0cXlNyT1Lb5bZrZO6MdCztkL9uqOBW5lisn7ygxnageyYTiybimdk7Juew0vS8AAADGlznsDgAAAAAA0E7uUk7xrXElphOKb43LjJuqOBXZy7Y821Pg35lVMmHIjJky46bMhKlYIqZYIibFJGOp90tqAQAAYLhIcgAAAADAJnPvvfcql8spl8vpvffeWzt/8eLFkf382ouvSYZkmEZ1k/Giq+L1ovLZvKyblpyCI8/25Hu+Aj+QgjtLX5mqJj4MU8q2CQwAAADGDstVAQAAAMAm8vTTTyubzX5qz42aUf5cPSH5ri+3UN1kPDYVk2u5MgxDnu2pvFqWW3Tl2Z4qbqWa8KgE1aQHAAAANiSSHAAAAACwyWQymYbnjx07NrqfHzum97/1vpyCo9JySZM3JmUYhpy8I2Oiutm4W3RVXi3LKTpyrbpkRxAoyJDsAAAA2GhIcgAAAAAARl56MS3XcmUv27JuWopNxuR7vhLTCZkxU4EfyLM9OUVH5ZWynMKdRIflVffs8AIpNeynAAAAQK+R5AAAAAAAjIUDjx7QlVevVBMbE9WNxxPTCZlxUwqkiluRa7lyCtVER21Wh71iSw8Mu/cAAADoB5IcAAAAAICxcPLlk3p28lnlc3lJkmd7SiQTMhOmDKO6ZJVne3Kt6rJV5ZVyNcHhSzohidWqAAAANhxz2B0AAAAAMB4Mw1g7opZrdm39+XbXo/YLG8OZ18/ILbjKX8srn81rNbuqQragfC6vwlJBxRtFWTcslW6XZN22FASBnnjjiWF3GwAAAH1CkgMAAABAW7UEQhAEn/p3J+VaXQuC4FNH/T3XX6u/Z7Nr2JgyRzI6/d3TKtwsaPmDZeWz1WRHIVeoJjk+Kqp4o6h8Li+34OrxVx9XapHNOAAAADYqlqsCAAAAEEot+dAumdCqXJh71BIXPRdIH1z8QO9/731d+8E1SdJd/+Vd2vcL+/TZhz8rkR8ZG/MPzeup8lO6cOqC3nnpHcWmYgr8O8kz05Bnezp46qCOf/v4kHsKAACAfiPJAQAAAGCk1SdE1idAWl1b7yd/+RP90ak/kr1syzANJbYmFPiBrv3gmn7wr36gqV1T+vrLX1fmi5n+PQh67sSLJ3TixRNaemtJ2TezkqozPVKHmLkBAACwWZDkAAAAADAyGs3iqD+3/t+trtX85f/5l/qL/+0vNPOzM9pzcI8S0wkZhiGv7On/Z+/ug+S47zu/f7rnYZc7u8ACiyVmZiks6ZAgKQgmsARoymcIcMwTr67MOAeAskhWHc/g2bHjB0ZOKn+owpKZsBxf5LuEFdsp23WEbEOkRAj0Hz6fJTMpGhYPVmRCNEUavlDCmfABs4tHLjDP3T3d+aMxy8VoHnp6nnffr6opzk73r/vX3+5hkd/v/H4/K2upuFxU/mJeR//Lo3rk1x/Rjzz7Iz28QvRCcndSyd0UNgAAANYjihwAAAAA1qyT//NJfevffEt3/Ogd2nz3Zk0mJxW7LSbXcVXOllW8VtTYlTHFbospeltUb/xPb8gu2IPuNnosyOifVvu1Okaj7UHPDQAAgGAocgAAAAAIpDpKotXi3s32C3qMblj++2V983/9prb96DZt/eGtmv34rDbMbZARMWRlLRWuFhRPxBWJRSRJnuvJdVz9xfN/If2ipKmedxEDUH32qs9hozVgmu3XagRRs2MG2Q8AAADBUeQAAAAA0NLqRG/176rVidpm+zXbVnuceude/XeQOkOdCAAAIABJREFUbX/8z/9Y0x+b1vRd05rZPqPkA0lt+qFNsnKWchdzMqKGvIqnilWRU3LkFB3ZeVvWsqXCawXp6fbjhNFQ+7y2u1+r0R+NtocpaCyeXlTm9M31RvaklVpI1d1v/sW2Dy1JOvds67bnnm28rVHbZm2CHKOd9u0eu53jh+1bbbt+n69X5+zG/W50vFbHaDemnTzXjdr34l50+xkNG5ew19buMQCgFyhyAAAAAAgkaOK2WSK3X9usnKXz3zqvbfu2aWLLhCZmJ5S4PaHxTeOSIcVyMcUn4opNxD563RZTdCKq2IaY9A+S4fR+tAnWpm5MSXX88HGdOXFGsYmYZEi6eRi7YGvH4zt0+NXDXehpMPMv1k9eNkt4VreFSe42O2c7etW/Zn0LW2zqJBZh+hnmnJ3Gs53zh9mvm7od0158h8LEpdv3EACGBUUOAAAAAGvO4ulFRcYifuFiLCpDhsq5svIX86pYFbm26yefDckwDRkRQ2bMVCQWUSQWkREx5F5wB30ZGAH1Rm6Embqq6vyp8zq6/6gSWxLatm+b4pNxGaYh13Zl5SyVlks6+/pZvTD2go68eUTpvWlJrROhQRKXzZKwzZK7YdsFad+pVnEJknxevT3odVW3tXsdjQoLvepnvXPWHrfVvu2eL6xuxbSVsDEN267TmIaJSy++swAwSOagOwAAAABgeJ07d07f/e539f777+v999/XxYsX9Qd/8Acr24f1/cXvXpRpmpIhVeyKylm/wJHNZJW/klf5ell2wfYLHhVXnuvdUvQwTVPGRUZyoLmwa2o0K3C8/NjLSi2kdNcjd2nuoTml96SV3JXU7P2z2nzP5pXp1xJbEzr26DFd+PaFblxKQ62Sq+0UPlZrVXzpRhI1SGK43WMESf6GTaq3kwBvtV8v4t+NeAY9f+2xB5FUbzemYdp1EtMwcenkOwsAw4wiBwAAAICGvvSlL+nkyZNKpVJKpVLauHGj9u/fv7J9WN9PbJmQW3HllBxZWUvFq0XllnJ+kWMpr8LVgsrXy7Jylpyi89HoDseTV/ELHt4EC0KvVdXppJqtx9Fqv24XOCTp6P6j2nz3ZqUWUkrvSeuOh+9Q6sGUZj8+q013b9L0/LQ2zG3QZHJSU6kpxW6L6aV9L7XdBzQXtADQ72mUas83rP0MahiT6mFjOur3AgBGHdNVAQAAAGjq6tWrmpqaWvn7zjvvHPr36T1peZ7nFziuFRWfjPt/T1n+VFSOK6foqHyjrHK2LDtv+4uPlx1V7Ipc15Uxx0iOtai6iHi1cNFoaqlW+63+5+rtzRYpb9bu+OHjSswmNLN9Rlvu26LkQlLT89OqWBXlL+UVHY/KMPxnt2JV5JQc2QVbpRslvfbZ13TwKwe7EJ3+YO7/wRp0/AddHOK56xwxBTBsKHIAAAAAWHOm75pWfDKu0rWSClcKisQjch1X5WxZkWhEnuvJKTuy8/ZKocPKW7KLtuyiLY1Jmh70VaBXGo2kaLa2RpDPO2l35sQZbdu3TYmtCU3NTWl6flobt21UabmkilWRXbBlZS3FJ+P+KxFXLBHT+KZxvfvquz0rcnS6kHaQ5OcgEqb8ov4j3Yh/u4uIDzIpPgx9CGJQxaBO7v+wxxTA2kWRAwAAAMCaY0ZN7f2Fvfqrf/NXik3GZJiGKlZFsURMkXhE8vy1OpyiIytnqXy9rPINf/oqO2dLn5Q8g+mq0B+LpxcVm4gpPhlXbCImM2aqYlVUWi7JKTny3JujSiKGzKgpM2bKjJuKxqP+azyqpbeXlNyd7KgfzZKqQdd46LTN6s/7kTAdhqTsuWf9a6634HMvE929iH/YBe57pZ/x7JVexSvMd7bePqMYUwBrD2tyAAAAAFiTDjx/QBNbJnTj/A3lFv31OKqv3FJO+Yt55S/763MUPyyqvFxW8VpRU3NT0qcG3Xv0wr333qvFxcWVV9Xqz1Z/fvLkyb68z5zOrCx677me7Lyt/KW8spmsClcK/toxJUeu48pzPcm7OeWVKRkRv03mrUzouPRK0ORndTHkfi0wPcy/Oq8WO6qvfug0/kHj2e9keKvF04c5OT+oZ7RVTEY5pgDWNkZyAAAAAFizfuG9X9Bvbf8tLX+wrEQxsTKSo5pMrlj+aA47b6twraDx6XH9/Ds/r1//4q8Puuvosi984QvKZDJ1p4wKMr1Ur9/Lk1zblZ2zVbxWVHQ8KrtgyzAMOSV//Zjq2jEVu+IXPCqeX/TokkYJ1UajDFq1Dduu2ciGTg1rgaPRwtS9XrC60/i3W+AYhrhXr29Y9StWYb+zjY41zDEFsPZR5AAAAACwZsUTcX3uHz6n3/+R39e1712TYRp+oSMaked5ci1XVsGSPOn2HbfrX37rX0qsN75mpdPptj4/cOBAX96n9/jnt3KWistFjV0ek2EYsrKWjIi/2Hh1/Rgrb8ku3FrsMExD6b31r6EbwhYdelmsCGOYEu2NDHPfaoWJZ7NE+Cjcn14bdAyG7TsLAEFR5AAAAACwphkRQz/31s/pnS+9o1P/+pSuvn9VZsyfude1Xc3cO6Mf/R9+VA/88wcG3FOsV6mFlOyCrdJySYUrBUXHonIdV/HJuMyoKc/15JQcWXl//Rgrd7PQUXDklPxXcldn63GsdYNOHndiGPs+jH0adcQUAMKjyAEAAABgXXjgXzygB/6FX8i49r1rkqTN92weZJeAFTse36Gzr5/1CxsRf+Hx+GTcL8h5UsWuyC7YsnJ+oaM6qqN0vaSdT+wcdPfb0u+pbdpNHrf6NfuwJKPD9rPT+Ie5/mb79jueza6905iGfWaG5ZkKi6mqAAwaC48DAAAAWHc237OZAgeGyuFXD8su2MouZpVbyimbySp7Iatsxv87fzGvwuWCileLKl4rqny9rNL1kuRKB798sKd9a5TAbJXYDJL4rLdPNxO+nR6rtn+9Sua2inG7i3oH7We78R+VZHyQZ7ZfMQ36PelHTDv5znYjpgDQK4zkAAAAAABgCBx584iOPXpM2fNZ2QVbsURM0XhUMiWv4qliV+QUnJURHJ7n6ZlTz3Tt/K0SoM2S3u22Wz2aoB+Fg1a/3q/3WbP+tfpFfqvPw8ayXj/C9DNM/DuJZyfCxjTsc9WrmLa65+3GtJdxaff5BoBBYyQHAAAAAABDIL03rae+/pRyV3Ja/mDZH82RySq3mFsZzZG/nFd2MSs7Z+vp159WcqH3a3Gce7ZxISBMu1btW7Xrl2b96+e5gsS5nc+7cc5h1+q5G0RMh0En39lOYwoAvcRIDgAAAAAAhsTcQ3N6rvycTjxxQu999T1Fx6PyXE+SZJiGnJKjnU/sDDxFVZDEYyfJyU4Tm71MjHbj2O0eY1CxDNu2W+tqdCJs4aDT4/aqfb9iOoi4UMgAMKwocgAAAAAAMGQOvXJIh145pKW3l5R5KyPJH+mR3NX7kRsAAACjhCIHAAAAAABDKrk7qeRuChsAAACNUOQAAAAAAABDr51Fj5lWBwCA9YOFxwEAAAAAAAAAwEhiJAcAAAAAABh6jM4AAAD1MJIDAAAAAAAAAACMJIocAAAAAAAAAABgJFHkAAAAAAAAAAAAI4kiBwAAAAAAAAAAGEkUOQAAAAAAAAAAwEiiyAEAAAAAAAAAAEYSRQ4AAAAAAAAAADCSKHIAAAAAAAAAAICRRJEDAAAAAAAAAACMJIocAAAAAAAAAABgJFHkAAAAAAAAAAAAI4kiBwAAAAAAAAAAGEkUOQAAAAAAAAAAwEiiyAEAAAAAAAAAAEYSRQ4AAAAAAAAAADCSKHIAAAAAAAAAAICRFB10B9YKwzBW3nue1/Z+zdqv3tZqe7NzAwAAAAAAAACwljCSowuqRYZqgaG2KBFkP8/zmhYoqtvrFThWb2t0bgAAAAAAAAAA1hpGcnRJtfjQqtAQdL9eWfqbJZ37i3M69+Y5SdL8p+Y1/6l5JXcl+94XAAAAYBR0c9R2vWO0Oj4jtwEAAIDGKHKMiE7/x+bKf7yir/7XX9WN8zckQ4reFpXnevr+v/++DNPQ1Mem9OSfPKnNd2/uZrcBAACAkbZ6NLZhGCsjqdvdr9F/w9fu1+pvAAAAALeiyDECGk1RVd3WakTI6d89ra9/7uuanp/Wx37sY4pPxmUYhipWRVbWUnG5qNxSTr+z83f0k7/zk9r1M7t6di0AAADAqBnUqO0wBY7F04vKnM5IktJ70kotpOruN/9iuD6de7Z123PPtj5O7TGCtGl2jDDtWx1ztaDHr9e+l/Ho1vl6dc5O49nseK2O0W5MO32u+3Uvuv2M9rJdt+8/AACNUORYA5oNhf/2b31bbzz3hj72yY9p892bNZmcVPS2qFzHlZWzVLxW1NiVMcUmYopfjOvPnv0zOZbT70sAAAAA1rSwI7ODtjt++LjOnDij2ERMMiTd3NUu2Nrx+A4dfvVw230Oa/7F9pPeYc7Rbc2OWd0WJvnci3i06mu3+xnmnJ3Gs53zh9mvm7od00btOolpL561sO3C3H8AAJqhyNEl1V9ZtfrFVtD9OulDVf5yXn/+3/+5PvajH9PWH96q2Y/PampuSmbElJWzVLhaUDwRVyQe8du6nlzH1Z/98p9JvyppvOtdBAAAANadTkZmt5q66vyp8zq6/6gSWxLatm+bP2rbNOTa/o+aSsslnX39rF4Ye0FH3jyi9N60pNaJ0CDJx2ZJ2FbJ3eq2QSSkm2kVlyDJ59Xbex2PRoWFXvWz3jlrj9tq33bPF1a3YtpK2JiGbddpTDuNSzfi2Y/7DwBYX8xBd2AtqP6Pxup5eKvq/fKq0X7Vz1e/X/13vbl9G30uSV/76a9p6o4pTd81rZl7Z7R111bNPTSnLfdv0YaPbdBkclITsxO6bfNtGt80rvHpcY1tHFN8Ii4d70poAAAAALTged7Kqx3nT53Xy4+9rNRCSnc9cpfmHppTek9ayV1Jzd4/q833bPb/X2D7jBJbEzr26DFd+PaFHl2FL0iCvNOkZq9+BR4kMdzuMXoRj2bX36t+top5u33p5jXXO/YgEuftxjRMu05iGjYuvWhHYQMA0G0UObqk0f+Y1Pu70X61r0bbghzPKTo695fnNL1tWonbE0rcntBUckoTWyY0tmFM8am4YomY/5q49TW2cUz6e0mVLgQGAAAAGHGrf4zUjf2atW+n0HF0/1FtvnuzUgsppfekdcfDdyj1YEqzH5/Vprs3aXp+Whvm/B83TaWmFLstppf2vRSqb8NmmJKkQQsA/R61Unu+Ye1nUMN0z6vCxnTU7wUAAMOG6arWqMxbGUXHo4pNxBQdi8owDFl5S/nLeVXKFbm2K3n+/0iZEVNm1FQkFvFf8YiMqCFlBn0VAAAAwGBVp5NqNBq7dtqpVqO7m22r94OmRtuPHz6uxGxCM9tntOW+LUouJDU9P62KVVH+Ul7Rcf//AVzHVcWqyCk5sgu2SjdKeu2zr+ngVw6GjskgkfTtv0GvnzDo4tAwFlcAAMCtGMnRgXQ6rU996lP6/ve/v/Kq+v73v69vfvObK3+/8sorfX1/8Z2LMk1TMqSKXZGVs5S/lFc2k1XhSkHlG2U5RUcVqyK34kqu/MUJDckwDb/tUoigAAAAAGtMt0dtB2nTavuZE2e06e5NSmxNaGpuStPz09q4baMmtkz409BuGFN8Mv7RK+GP5B7fNK53X303bCha6mVCepDJdoorH5l/8dZX2GO0s98gCw2dXGc/jUIfq4bhvgIA1hZGcnTgZ3/2Z3X9+vW622ZnZzU2Nrby94MPPtjX9+W/LsutuKqUKyuLjEfiEVl5S4ZhqFKuqJwty87bcoqOHMtRxanIq3jy3Jv/E8XC4wAAAMDQWTy9qNhETPHJuGITMZkxUxWrotJySU7JkefeHFUSMWRGTZkxU2bcVDQe9V/jUS29vaTk7mRH/WiWVO31YtKDMujzV/tQTbw3Wni8Fxodu5OEddgF7nuln/HslWF4RmvVi+Mw9hMAMLoocnRo48aNDT9fvW379u19fX/FvSLP81TOllW8VlR8Mi5JimfjMqOmXMeVXbBVvlGWlbNkF2w5JUdO2R/d4XmevHR7Cx8CAAAAa8m9996rxcXFlb9TqZQk3fJZtz6/dOmSHnjgAUnSyZMntX///obvM6czKyOwPdeTnbeVv5T3/zve9WTlLDklR67jynO9lWlqZUpGxG+TeSvTcZGjnwaZbB7mX50PIi6dFgKCxrPf19Zo8fRqP+oVlYbFMD+jjQxzPAEAo4cixxo1c8+MItGISh+WVLxSVHQsKtdxFZ+MKxKLyHM9OSVHVt7yCx1ZS3bu5qiOkiPP9KTNg74KAAAAYDC+8IUvKJPJ1J1GqtnUUmE/b/e9PMm1Xdk5W8VrRUXHo7ILtgzDkFNyVL5xc9R2yVHFrvgFj5ujtrulUYKy0SiDsIZhmqphS8Y2Wpi61wtWNysEBLnn7RY4hiHuqwsdw2iYYtVIvb51+98TAID1jSLHGmVEDC383IK+83vfUXwyLsM05JSdlSKHJH8BwqKjcq6s8vWyylm/2GFlLWmvJKP5OQAAAIC1LJ1O9+3z1e8PHDjQ9H16j7+vlbNUXC5q7PKYDMOQlbVkRPzFxu38zVHb+Y9GbVeLHYZpKL23fp+6oZ2kdzuaJZp7kegd1eTxsAoTz37f81EzyjHo1b8nAADrE0WONezTv/lpvfvyu7rxn2/I8/yRG7FETJF4RDIkz/HklB1/2qqsX+gofVjSbTO3yX7E9hciBwAAADBUUgsp2QVbpeWSClcKt4zaNqPmraO2r6+anrbgj9p2So6Su0ZnqqpBGOXk8TD2fRj7NOqIKQAAH6HIscb94plf1O/c/zu6fu66nMJHRY7q/L3V0RxW3lLxWlFjU2P6hfd+Qf/q//hXg+46AAAAgAZ2PL5DZ18/6xc2Iv7C4/HJuMyYKXlSxa7ILtiycn6hozqqo3S9pJ1P7Bx099vSLIk7DCM4Wv0ifViS0WH72el0TWGuv9/3vJlm195pTMM+M8PyTAEAMCzMQXcAvTU+Pa5f+t4vaWLLhLKZrK6fu65sJqvsYla5pZzyF/PKLmZVvFbUxm0b9cvf/2WNbxwfdLcBAAAANHH41cOyC/bKf9dnM1llL2SVzXz03/mFywUVrxZVvFb0R21fL0mudPDLB3vat2Fev6CVTpPHtdfeq1g0Om7YRb2D9rPefs3OOSrJ+FbxlPoX01btRj2mQbcDANAORnKsA/HJuH7p//slfet//5a+9eK3dP3cdcUn45L8eXyn75zWw597WD/yKz8y4J4CAAAACOrIm0d07NFjyp7Pyi7YiiViisajkil5FU8VuyKn4KyM4PA8T8+ceqZr52+VpGyW9G71+SAXGa99X6vZ4tuN2rb6RX6rz9uJZTNh+9mqXT2dxLMTYWMaNuneq5i2uuftxrTbcenWMzrsxRoAwGigyLGOPPy5h/Xw5x6WJC1+Z1GSP58vAAAAgNGT3pvWU19/Si/te0mlGyWNbxpXNB6VEfGnpnUdV07JWRnB8cypZ5Rc6P1aHOs1adloWqdexKOTc4Vt28/r67dmU3IR03BaTXO2Fq4RADA8KHKsUxQ3AAAAgNE399Ccnis/pxNPnNB7X31P0fGoPNeTJBmmIafkaOcTOwNPURU0oRtWtxOb3TxeN47V7jEGFcuwbbu1rkYnghQOenHcXrXvV0wHERcKGQCAfqHIAQAAAAAj7tArh3TolUNaentJmbcykvyRHsldvR+5AQAAAAwSRQ4AAAAAWCOSu5NK7qawAQAAgPWDIgcAAAAAYM1pZyFpptUBAAAYXeagOwAAAAAAAAAAABAGIzkAAAAAAGsOozMAAADWB0ZyAAAAAAAAAACAkUSRAwAAAAAAAAAAjCSKHAAAAAAAAAAAYCRR5AAAAAAAAAAAACOJIgcAAAAAAAAAABhJFDkAAAAAAAAAAMBIosgBAAAAAAAAAABGEkUOAAAAAAAAAAAwkihyAAAAAAAAAACAkUSRAwAAAAAAAAAAjCSKHAAAAAAAAAAAYCRR5AAAAAAAAAAAACOJIgcAAAAAAAAAABhJFDkAAAAAAAAAAMBIosgBAAAAAAAAAABGEkUOAAAAAAAAAAAwkihyAAAAAAAAAACAkUSRAwAAAAAAAAAAjKTooDsAAAAAAEC3GYax8t7zvFD7tTpGkO3Nzg0AAIDOMZIDAAAAALCmVIsP1QLD6mJE0P2qBYrqq/YYtdsbHRsAAAC9xUgOAAAAAMCaUy081CtQBNmv1eiPINuDFjoWTy8qczojSUrvSSu1kKq73/yLgQ73A84927rtuWfbO+bq4wVpW+/87Z6znWO3c/ywfatt1+/z9eqcncaz2fFaHaPdmHb6XPfrXnT7GQ1zL4Leh27/uwIA+oEiBwAAAAAAbWpUEGlniqrjh4/rzIkzik3EJEPSzWZ2wdaOx3fo8KuHu9nlpuZf7CxhHmbfds4Z9vhSuIRus76FLTZ1Eosw/Qxzzk7j2c75w+zXTd2OaaN2ncS0W3EZRHwBoJ8ocgAAAAAA0ES9wkW9wkbQAsf5U+d1dP9RJbYktG3fNsUn4zJMQ67tyspZKi2XdPb1s3ph7AUdefOI0nvTklonQoMkn5slYbtRdAhz3l4ce/XxgySfV28PGo/qtnavo1FhoVf9rHfO2uO22rfd84XVrZi2EjamYdt1GtOwcekEIzYAjBLW5AAAAAAAoIF2Fw83DGPlVf17tfOnzuvlx15WaiGlux65S3MPzSm9J63krqRm75/V5ns2a/quac1sn1Fia0LHHj2mC9++0NVrqhXml/ntTOnTaN9uJFGDJIbbPUaQgkHYpHo7CfBW+wWdiqmd+HcjnkHPX3vsQSTV241pmHadxLQbcQkzAgcARg1FDgAAAADAmtOoyNDOfu0WOFYvRL56rY/Vju4/qs13b1ZqIaX0nrTuePgOpR5Mafbjs9p09yZNz09rw9wGTSYnNZWaUuy2mF7a91LgPvTDWkiWBi0A9Huan9rzDWs/gxrGZyVsTEf5XgzjfQCAbmK6KgAAAADAmlKdOqpauGi0Zkar/Vb/c/X2dhYpX+344eNKzCY0s31GW+7bouRCUtPz06pYFeUv5RUdj8owDLmOq4pVkVNyZBdslW6U9NpnX9PBrxwME46uCbMWB8nVwRh0/AddHOK583VyH4gpgFFCkQMAAAAAsOY0Kjw0W1sjyOdBt9fb58yJM9q2b5sSWxOampvS9Py0Nm7bqNJySRWrIrtgy8paik/G/VcirlgipvFN43r31Xd7VuQIkgjtNGk+iITpMP6iflC6Ef92FxEfZFJ8GPoQRC+f0W7HYFRiCmB9osgBAAAAAECPLZ5eVGwipvhkXLGJmMyYqYpVUWm5JKfkyHNvjiqJGDKjpsyYKTNuKhqP+q/xqJbeXlJyd7KjfjRLqoZZvDrs+fqZMB2GpOy5Z/1rrrfAdD8S3Y0+DxObsAvc90o/49krvV7IvdN2oxhTAOsLa3IAAAAAANaMxcXFW16tPn/zzTdX3p88ebJn7zOnM5IhGaYhz/Vk523lL+WVzWRVuFKQlbPklBy5jivP9STv5lRZpmRE/DaZtzKh49KJbiQ4qwso92uB6WH+1Xm12FF99UOn8Q8az34nw1stnj7MyflePqOdXPcoxxTA+kWRAwAAAACwJjz//PN64403fmDxb6n+ouDVz/v1Xp7k2q7snK3itaJySzllM1nlL+ZVvFpU+UZZdt6WU3JUsSt+waPi+UWPLqlNdq9OetdLXnYjEdssadqLhOmwFjgaFRh6XfjpNP7tFjiGIe7D0Idm+lHg6Paxhz2mANY3pqsCAAAAAKwZ77//vp588skf+DydTtfdf9++fSvvDxw40LP36T3++a2cpeJyUWOXx2QYhqysJSPiLzZu522Vb5Rl5S3ZhVuLHYZpKL23/jV0Q7PplKTmyfBhSm4PU18aGea+1QoTz1F5VgalXzHgPgBYTyhyAAAAAADQY6mFlOyCrdJySYUrBUXHonIdV/HJuMyoKc/15JQcWXlL5etlWbmbhY6CI6fkv5K7OluPY60b5cTtMPZ9GPs06ogpAPQGRQ4AAAAAAPpgx+M7dPb1s35hI+IvPB6fjMuMmZInVeyK7IItK+cXOqqjOkrXS9r5xM6B9LlZMrZZwrY6MqRf2k0eBx25MuhkdNh+dhr/MNcf9lnphWbX3mlMwz4z/YpBr+4Da3EAGGasyQEAAAAAQB8cfvWw7IKt7GJ2ZT2O7IWsshn/7/zFvAqXCypeLap4rajy9bJK10uSKx388sGe9q2XCcxerfXRrWPV9q9XsWh03LCLegftZ7vxH5YCTyut4in1L6at2q2nmALAIDCSAwAAAACAPjny5hEde/SYsuezsgu2YomYovGoZEpexVPFrsgpOCsjODzP0zOnnuna+VslY7uZwFw9mqAfhYNWv96v91mz/rX6RX6rz9td26SRsP0ME/9O4tmJsDEN+1z1Kqat7nm7Me3kWQuLERsARhEjOQAAAAAA6JP03rSe+vpTyl3JafmDZX80Ryar3GJuZTRH/nJe2cWs7Jytp19/WsmF3q/Fce7Z3vxCu1myeBh+Ed6sf/08V6vzhe3nsMe/E836P6iYjrpOYwoAg8JIDgAAAAAA+mjuoTk9V35OJ544ofe++p6i41F5ridJMkxDTsnRzid2Bp6iKkjisZ9J+36cu5vHbvcYnZxzEG27ta5GJ8IWDjo9bq/a9yum3b4fg/p3BQD0GkUOAAAAAAAG4NArh3TolUNaentJmbcykvyRHsldvR+5AQAAsFZQ5AAAAAAAYICSu5NK7qawAQAAEAZFDgAAAABYpwzDWHnveV6o/Rpta3XsoOfG+tLOosdMqwMAACQWHgcAAACAdalaZKgWGFYXHYLuZxiGPM9bedUWLhoVL5q1AwAAANrBSA4AAAAAWKeqRYhWhYZG+4UdgRGm3eL/xE0oAAAgAElEQVTpRWVO31y3Yk9aqYVUqHNjuDE6AwAAtIsiBwAAAACg74IWS44fPq4zJ84oNhGTDEk3d7ULtnY8vkOHXz3c454CAABgmFHkAAAAAAB0rDoFVVC163fUtj1/6ryO7j+qxJaEtu3bpvhkXIZpyLVdWTlLpeWSzr5+Vi+MvaAjbx5Rem+6a9cCAACA0UGRAwAAAADQkXYLHK2cP3VeLz/2slILKc1sn1Fia0KxiZg815Ods1VcLqpwpaD4ZFzZxayOPXpMT339qa6dHwAAAKODIgcAAAAArFPV4kSrhb+b7RemwNGqzdH9R5VaSCm1kNKW+7Zoam5KZsyUnbdVvFbU2OUxRceiMiOmJCl7PquX9r0kfb6tbgAAAGANoMgBAAAAAOtQtWhRLVw0mj6q1X6r/7l6++rPats2W8D8+OHjSswmNLN9Rlvu26LkQlLT89OqWBXlL+UVHY/KMAy5jquKVZFTcmQXbJVulGQdtySW6AAAAFhXKHIAAAAAwDrVaDRF7edB9wu6rdn2MyfOaNu+bUpsTWhqbkrT89PauG2jSsslVayK7IItK2spPhn3X4m4YomYxjeNyzpjNT0nAAAA1h5z0B0AAAAAAECSFk8vKjYRU3wyrthETGbMVMWqqLRcklNy5Lk3R5VEDJlRU2bMlBk3FY1HFY1HpahkLDWfegsAAABrC0UOAAAAAFhnFhcXb3nV+/zq1as6efLkyrZ+vM+czkiGZJiGv8h43lb+Ul7ZTFaFKwVZOUtOyZHruPJcT/JuToVlyi98GKaU6SQyAAAAGDVMVwUAAAAA68jzzz+ve+65RwcOHPiBbc2mqerbe09ybVd2zl9kPDoelV2wZRiGnJKj8o2y7Lwtp+SoYlf8gkfF84seAAAAWHcocgAAAADAOvP+++/rySef/IHP0+n0LX+vLoT04/3iBn9UiZWzVFwuauzymAzDkJW1ZET8xcbtvK3yjbKsvCW7UFPs8Dx5aYodAAAA6wlFDgAAAADAUEgtpGQXbJWWSypcKSg6FpXruIpPxmVGTXmuJ6fkyMpbKl8vy8rdLHQUHH/NDseTkoO+CgAAAPQTRQ4AAAAAwNDY8fgOnX39rF/YiPgLj8cn4zJjpuRJFbsiu2DLyvmFjuqojtL1kvSJQfceAAAA/UaRAwAAAAAwNA6/elgvjL2g7GJWkuSUHMUTcZlxU4bhT1nllBzZBX/aqvL1sl/gcCUdksRsVQAAAOuKOegOAAAAAACw2pE3j8jO2cqezyqbyepG5oZymZyyi1nllnLKX86rcLmg4rWiCtcK8jxPz5x6ZtDdBgAAwABQ5AAAAAAADJX03rSe+vpTyl3JafmDZWUzfrEjt5jzixwX88pfziu7mJWds/X0608rucBiHAAAAOsR01UBAAAAAIbO3ENzeq78nE48cULvffU9Rcej8lx/LirDNOSUHO18YqcOfvnggHsKAACAQaLIAQAAAAAYWodeOaRDrxzS0ttLyryVkeSP9EjuYuQGAAAAKHIAAAAAAEZAcndSyd0UNgAAAHArihwAAAAAgDXHMIyV957ntb1fq/ZB2gU5PwAAADrDwuMAAAAAgDWlWmioFhfqFR5a7ed5XsPihGEYK9s9z6vbrll7AAAAdA8jOQAAAAAAa061wFBbhAi7XxjVYkgri6cXlTl9c72RPWmlFlJ195t/MVw/zj3buu25Z9s75urj1WsbtK/tnrdVX8Icu177IG1r2/X7fL06Z6fxbHa8VsdoN6adPtf9uhfdfkbD3It27kOj84Y9NwD0GkUOAAAAAADaUG/0RhjHDx/XmRNnFJuISYakm4exC7Z2PL5Dh1893IXeBjP/YmeJz0Fp1pfqtjDJ52bxCHv9rfra7X6GOWen8Wzn/GH266Zux7RRu05i2q24tHucTp5VABgEihwAAAAAALShdoRGvREbzUZxnD91Xkf3H1ViS0Lb9m1TfDIuwzTk2q6snKXScklnXz+rF8Ze0JE3jyi9Ny2pdSI0SOKxWRK2m8nLIEniXp+r1XU1+mV70HhUt7WbQG5UWOhVP+uds/a4rfZt93xhdSumrYSNadh2ncY0bFw61ckoLQDoJ9bkAAAAAACgT86fOq+XH3tZqYWU7nrkLs09NKf0nrSSu5KavX9Wm+/ZrOm7pjWzfUaJrQkde/SYLnz7Qk/7FOaX+cPyS+4gieF2jxGkYBA2qd5OArzVfkGnYmq0X7t96eY11zv2IJ6pdmMapl0nMe1GXNr9zoZ5bgBg0ChyAAAAAADWnOp0Uq3W2Qi6X7cc3X9Um+/erNRCSuk9ad3x8B1KPZjS7MdntenuTZqen9aGuQ2aTE5qKjWl2G0xvbTvpb70LahOkpzD8ivwoIncfve39nzD2s+ghjEhHjamo3wvhvE+AEA3MV0VAAAAAGBNqa6ZUS1cNJpaqtV+te8bLVIedKqq44ePKzGb0Mz2GW25b4uSC0lNz0+rYlWUv5RXdDwqwzDkOq4qVkVOyZFdsFW6UdJrn31NB79ysKO4dKqbyVuSrr016BE3gy4O8Xz5wq7FQfwAjBqKHAAAAACANafRehi1nwfdr53tjbadOXFG2/ZtU2JrQlNzU5qen9bGbRtVWi6pYlVkF2xZWUvxybj/SsQVS8Q0vmlc7776bs+KHEESoaOY/BzGX9QPSjeKAO0uIj7IZ2UY+hBEL5/RbsSA4hGAUUGRAwAAAACAHls8vajYREzxybhiEzGZMVMVq6LScklOyZHn3hxVEjFkRk2ZMVNm3FQ0HvVf41Etvb2k5O5kR/1ollQNs3h1mHP3O1E6DInZc8/6119vgel+JLobfR4mNmEXuO+VfsazV3q9kHu7evHcAEAvsSYHAAAAAGDNuPfee7W4uLjyqqr+fePGDZ08eXLl8369z5zOSIZkmIY815Odt5W/lFc2k1XhSkFWzpJTcuQ6rjzXk7yb02SZkhHx22TeyoSOSydGMWk8zMnYarGj+uqH6gLWYReyDhrPfj8rrRZPH+Znt5fPaLeuu9PnBgD6hSIHAAAAAGBN+MIXvqD9+/fL87yVV9Xqv2s/79d7eZJru7JztorXisot5ZTNZJW/mFfxalHlG2XZeVtOyVHFrvgFj4rnFz26pDZpuTp5WS8xOszFgkaGtc+NEsW9TiA3KwR0c6qyYYr7MPShmX4UODo9dqfPDQD0E9NVAQAAAADWjHQ63fLzAwcO9P19eo9/fitnqbhc1NjlMRmGIStryYj4i43beVvlG2VZeUt24dZih2EaSu+tf23d0Gw6Jal5UjNIUrVfCfBhSrQ3Msx9qxUmnp0+K2tdv78Lg+wDAPQLRQ4AAAAAAHostZCSXbBVWi6pcKWg6FhUruMqPhmXGTXluZ6ckiMrb6l8vSwrd7PQUXDklPxXcldn63GsdaOcuB3Gvg9jn0YdMQWA3qDIAQAAAABAH+x4fIfOvn7WL2xE/IXH45NxmTFT8qSKXZFdsGXl/EJHdVRH6XpJO5/YOZA+d2N0Rj+mtmk3eRx05Mqgk9Fh+1ltF1aY6x+GkTy156un05iGfWb6FYNO7kOnzw0ADAprcgAAAAAA0AeHXz0su2Aru5hdWY8jeyGrbMb/O38xr8LlgopXiypeK6p8vazS9ZLkSge/fLCnfetHYrNXyd1Ok8e1196rWDQ6bthiUdB+trvWyrAUeFppFU+pfzFt1W5UYrraWlmjB8D6wEgOAAAAAAD65MibR3Ts0WPKns/KLtiKJWKKxqOSKXkVTxW7IqfgrIzg8DxPz5x6pmvnb5WMHbXk5errafXr/XqfVdvUa9vqF/mtPm93bZNGwvazVbt6OolnJ8LGNGxBqlcxbXXP241pJ89aWGGeGwAYNEZyAAAAAADQJ+m9aT319aeUu5LT8gfL/miOTFa5xdzKaI785byyi1nZOVtPv/60kgu9X4vj3LO9KXAM+y+/myWz+3muVucL289OzjnsmvV/UDFdK9bycwNgbWIkBwAAAAAAfTT30JyeKz+nE0+c0HtffU/R8ag815MkGaYhp+Ro5xM7A09RFSTp2M+kfa/P2+3jt3uMTs45iLbdWlejE2ELB50et1ft+xXTbt+PYXgWAKAXKHIAAAAAADAAh145pEOvHNLS20vKvJWR5I/0SO7q/cgNAACAtYIiBwAAAAAAA5TcnVRyN4UNAACAMChyAAAAAACAodDOQsdMpwMAACQWHgcAAAAAAAAAACOKkRwAAAAAAGAoMDoDAAC0i5EcAAAAAAAAAABgJFHkAAAAAAAAAAAAI4kiBwAAAAAAAAAAGEkUOQAAAAAAAAAAwEiiyAEAAAAAAAAAAEYSRQ4AAAAAAAAAADCSKHIAAAAAAAAAAICRRJEDAAAAAAAAAACMJIocAAAAAAAAAABgJFHkAAAAAAAAAAAAIyk66A4AAAAAwDAzDGPlved5be/XrP3qba22Nzs3AAAAsF4xkgMAAAAAGqgWGaoFhtqiRJD9PM9rWqCobq9X4Fi9rdG5AQAAgPWMIgcAAAAANFEtPrQaSRF0PwAAAADdw3RVAAAAADBA3ZiS6tJ3L+lbL35L//DmP2j5g2VJ0vSd09q2b5s++d99UrOfmO1KXwEAAIBhQ5EDAAAAAAak0RRV1W1Bpqj6d//Nv9OZr52RYRoa3zSu2z9xu9yKq9K1ks4cP6O/e+3vtPOJnfqnv/1Pe3INAAAAwCBR5AAAAACAIdVoAfOqf/vwv9W1719TcndSk8lJxRNxSZJdtFW+UVbxWlGFKwW984fv6OJ3L+pnvvkzfes7AAAA0A8UOQAAAACgieroilajKoLu10kfVvvSp76kDz/4UPMH5jWzfUZTqSlFxiKqWBWVr5dVuFpQfDKu6FhUZsTU0t8s6Q9/4g+lT3W9ewAAAMDAUOQAAAAAgAaqRYtq4aJ2ZEXt1FKN9qt9X91eWxBp1K62wPHdL39XS+8saX7/vG7/xO3aunOrNtyxQTKk0ocl5S/nFRmL+H10PVXsiipWRef/3/PSjKSPhw4JAAAAMFQocgAAAABAE40WA6/9POh+3dj2l8//pTb/0GZt/NhGbfqhTZrdMauZe2dUuFxQbiznFzbKFTlFR1bekpXzX2OTY7LfsNd9kSPoYu9B9qsdZRO2cAUAAIBwzEF3AAAAAAAQXPZCVsv/sKzbttym8U3jGt84rthETF7Fkxk1ZcZNRcYiK6/oePSj10RUWpaMXPen1BoVrUbTtLNfo7ae56286n0OAACA7mEkBwAAAADU4bruLX+bprnyueu6ikb9/52ybVuxWKxv7zNvZWRG/UKGYRqq2BUVPyz6fXNcOUVHruNKnp+EN0xDZsT0CyBRU4ZpyMus70R77TRjYfbr5Rosqy2eXlTmdEaSlN6TVmohVXe/+RfDHf/cs63bnnu29XHqHSNMuyBt2tHo2oKep9/X1a3z9eqcncaz2fFaHaPdmHb6XPfrXnT7Ge31s9ZpWwDoBUZyAAAAAF1UXZchyCLVjfYLu231PujM66+/ru985zt65513Vl5V77zzjv7kT/5k5e8vfvGLfX2//MGyzIi5MiVV+XpZ+aW8sheyyl/Kq7RckpWz5JQcVayKvIonz/VWih4RIyLvw/Vd5OhUvYXga7cH+fdAM8cPH9fzxvM6+qmj+savfkPf+Nw3dHTfUT1vPK+vfeZroY8bRrNk8fyLjbeHbdctnZ6/n9cVNo7Ntgdp107bbl930P378azUO2c7n4dt10lMB/GsNdun3/cIAFZjJAcAAADQJaunt6kmOOslQpvt12pbo4Wva4+Nzpw6dUqe5+nXfu3XfmDb7t27tXv37pW/P//5z/f1/ff+/fdWRmyUrpdUuFKQETFk5SwZEUMVqyIrZ6mcLcsu2ivFDtd25Tr+KBTNtBMNrNaqwFHvO9nOFFXnT53X0f1HldiS0LZ92xSfjMswDbm2KytnqbRc0tnXz+qFsRd05M0jSu9NS2r9y/sgv7Kut0+1/fyLzY9Ru63ddr1KkLaKS6P+NRph0OvrajeOnfaz3jlrj9tq33bPF1a3YtpK2JiGbddpTPv1rNWeI+zzBgC9wEgOAAAAoItWT28Tdr+gx6jVbjJVkuyCLbtgt9UGg5Xek5bruCrfKKt4tajcxZxyizllM1nlFnPKX8qreLWo0nJJ5RtlWXnLL3aUbxY7XFdKD/oqRlvtSI1uFRfPnzqvlx97WamFlO565C7NPTSn9J60kruSmr1/Vpvv2azpu6Y1s31Gia0JHXv0mC58+0JXzt1IkGl22kncrt7e62RoO0WZoPv14ro6iWOj/YJOxdRov3b70s1rrnfsQSTO241pmHadxHRQz1q9fSlsABg0ihwAAADAGtBOgePsN87qjz79R/ri7Bf1Gxt/Q7+x8Tf0xdkv6tijx/T3//ff97in6FTi9oS23LdFxctFFa4UlFvyCxzVV24pp/zlmkJHzvILWnlbmpU0MeirGKygxYl6+61eVDxsQbKRo/uPavPdm5VaSCm9J607Hr5DqQdTmv34rDbdvUnT89PaMLdBk8lJTaWmFLstppf2vdSVc/fSqE1jE7QAMOgplIa1n0ENY2I8bExH/V7UajZd2Vq5RgBrC9NVAQAAACOi0SLH7RQ4XnnsFZ3/q/OKjEU0lZ7S5ns2y3VclT4sKXM6o1c/86ru3H+nfvqPf7rb3UcXffo3P62v/LOvKDYZkwypUq4onojLjN9cHN125ZQcWXlL5etl/5Utyyk70k8NuPMDtnoquOrfVau/S832a6b2O1p7/Nr31e3HDx9XYjahme0z2nLfFiUXkpqen1bFqih/Ka/oeFSGYch1XFWsipySI7tgq3SjpNc++5oOfuVgu6HAOtXOKIpenn9Q5xvG4goAoDMUOQAAAIAR0iphuvrv1ft6rqffvv+3VVouKb03rcmtk36C3JPsoq3y9bKK1/yRAf/p//lP+u0dv61ffO8Xe39BCOWH/vEP6e5/crfOfuOsPM+TU3QUS8QUiUf8RHjFT4TbBdtfn+N6WaVrJW3/r7brb+/8W2mdrzveqGBR+3mQwkY7bZptO3PijLbt26bE1oSm5qY0PT+tjds2qrRc+uheZi3FJ+P+KxFXLBHT+KZxvfvquz0rcqzVX2Wv1esKoxtFgHYWEQ97jm4Zhj4EwTMKAMFR5AAAAAC6qFpcCDINTquRGc2OUfuL80bbqn534XdlF2zd+eN3amb7jKbSU4qOReWUHZWW/cWr44m4nyQ3DS1/sKzf3/v70mMBLxx995mvfUZf++mv6Xt/+j3ZBVuxREzReNSflNiVKvbNX/vnbZWzZd33z+7TwS8f1N8+/7eD7jpqLJ5eVGwipvhkXLGJmMyYqYpVUWm5JKfkyHNvjiqJGDKjpsyYKTNuKhqP+q/xqJbeXlJyd7KjfjRLqjaaw3/+xfqLDY9KgnYYEt2DimOjY3dSBAi7wH2vjOpzuVo347UWvrMAUA9FDgAAAKBLujENTqtjrN4vqG//n9/W8gfLuuvH79Ltn7hdW394qzbcsUHypOKHReUv5T8aAeC6fnK87Ojyf7wspSQ92H4s0B+Hv3pY/+F/+w/65q9/U/lL+ZXEtydPbsm/l9GxqH78f/lxffJXPzno7g7c4uLiLX+nUqmOPp+YmNDf/M3faP/+/ZKkkydPhnqfOZ2RDMkwDXmuJztvK38pr4pVked6snKWnJIj13HluZ7k3fz3gSkZEb9N5q1Mx0WOToxSgnSYf8k/iDh2muwOGs9+X1uzwly1P8P4DEj9eUZH6TsLAK1Q5AAAAAC6qBvT4AQ9RpD2XsXTqd88pZn/YkYb7tig6TunteX+LZq5Z0aFKwWZMVOu467M72/lLFlT/mtsckz2m7a0EOi0GJB/9D/+Iy0cWdDp3zuts39+Vpf/7rIMGbp9z+26+9N3a8/P79H4pvFBd3Pgnn/+ed1zzz06cODAD2xr9ztXW5jsxnt5/loqds5W8VpR0fGo7IItwzDklByVb5Rl5205JUcVu+IXPCqeX/TokkYJ1Ua//F7dptGUR8OWSB3WAseg4tjuCJ1a7RY4hiHuqwsdw6jXsRq17ywABEGRAwAAAFjDrv/n68pfymvLfVs0tnFMY1NjisajqpQrkiGZUVOReGTlFR2PrrxiiZh0VdKNQV8FWrlty236sc//mH7s8z826K4Mtffff19PPvnkD3yeTqfr7h/k89VFk7Dv03v841k5S8XlosYuj8kwDFlZS0bEX2zcztsq3yjLyluyC7cWOwzTUHpv/b52Q5Ck9zAkr1sZpkR7I8Pct1ph4tksgT4K96fX+hmD9RxnAGuPOegOAAAAAGtBIpFQuVxeeeVyuZVtH3744cDeL55elBExVtbaqFgVFa4WlM1kVfqwJKfoyLXdj34NfnPKHCNiyIyYMk1TyrQdDgBtSC2kZBfslfVxcks5ZTNZZTNZ5RZzyl/Mq3CloOKHRZWvl2XlbhY6Co6ckv9K7hrcVFWNDFPSepj60q5h7Psw9mnUDUNMh6EPABAGRQ4AAACgQwcPHtRP/MRP6Hvf+97K64033ljZ/kd/9EcDe3/jwg2ZkZtTUhX9RcbzF/PKZrLKX8yr+GHxo/n+bfeWOf8lyTRMRnIAfbDj8R3KZrIqXL61yJFdzCq3lFP+8q2FjuqojtL1knY+sXPQ3R9q7SZuW03bMyyJ4LD97LTfYa7/3LONX7X79Fqz0SSdxjTsMzMsz1Qzo/K9ALA+UeQAAAAAOrRz504tLCzoE5/4xMrrscceW9n+K7/yKwN7P3v/rD/VTcFW6br/K/Hs4s1fiC/lVpKmVtaSlfeLHZVyxZ8Gp+Kq4lVk3P7RgucAeuPwq4dlF+yVokY2k1X2wkff1fzFvAqXCypeLap4zS90lK6XJFc6+OWDPe1bkCmGGn0+6IRnp/2ovb5erVfQaRzD9rPefs3OOSz3tZVW8ZT6F9NW7fod0258Z/v1vQCAoFiTAwAAAFjD0nvSqtgVlW+UVbxWVGwiJrfiqjxVlhkx5bmenKKjcrYsK2vJztmyi/ZKscOreFLvpvoHsMqRN4/o2KPHlD2flV2wFUvEFI1HJVPyKp4qdkVOwVkZweF5np459UzXzt8qUdnqV+jdOFejxZDDWH2sIL/er/2s2qZe23ZjEeS6wsQxbD9btaunk3h2ImxMwybeexXTVve83Zj2+1mrHitMbACg1xjJAQAAAKxh45vGldyVVOFyYWUanNxiTtkL/lz/1WlwileLKn5YVOlGyS92FGzZeVtKSd6YN+jLANaF9N60nvr6U8pdyWn5g+Vb1uWojubIX84ru5iVnbP19OtPK7nQ+7U4mk0j1OzzUU949nOR9U7iGLaf6/HeVbcNIqbDotP7PgrXCGD9YSQHAAAAsMb95P/1k3rpUy8pfjEuSaqUK4olYorEI5Ik13bllPxfh5dvlP1Cxw1LdtmWnhhkz4H1Z+6hOT1Xfk4nnjih9776nqLjUX+dHEmGacgpOdr5xM7AU1R1krQMKkz7fiREu3GOdo/RyTkH0bbddTV6IWzhoNPj9qp9v2I6qLbdaA8A3UaRAwAAAFjjUg+m9MNP/bDee/k9eZ4np+SsFDkMw/DX3ihXZBdtWbmbhY7lknb/zG799e1/vbIIOYD+OfTKIR165ZCW3l5S5q2MJH+kR3JX70duAAAAjBKKHAAAAMA68NjvPaaxqTGd/t3TK3P9R+IRGaYhz/VWRnPYeVtWwdJDv/KQHvmNR/TXz//1oLsOrGvJ3Ukld1PYAAAAaIQiBwAAALBOfPpff1qpB1P60//2T1W8VpQMKRqPyvM8VcoVyZAisYh+6qWf0o7P7Bh0dwHgFu0slsx0OgAArB8UOQAAAIB1ZOeTO7Xj8R169+V3dfYbZ7X49qIkKbmQ1D2P3qNPPPkJmVFzwL0EAAAAgGAocgAAAADrjBkz9cDTD+iBpx8YdFcAIDBGZwAAgHr4iRYAAAAAAAAAABhJFDkAAAAAAAAAAMBIosgBAAAAAAAAAABGEkUOAAAAAAAAAAAwkihyAAAAAAAAAACAkUSRAwAAAAAAAAAAjCSKHAAAAAAAAAAAYCRR5AAAAAAAAAAAACOJIgcAAAAAAAAAABhJFDkAAAAAAAAAAMBIosgBAAAAAAAAAABGEkUOAAAAAAAAAAAwkihyAAAAAAAAAACAkUSRAwAAAAAAAAAAjCSKHAAAAAAAAAAAYCRR5AAAAAAAAAAAACOJIgcAAAAAAAAAABhJFDkAAAAAAAAAAMBIig66AwAAAAAAoDsMw1h573leqP1aHSPI9mbnBgAA6CZGcgAAAAAAsAZUiw/VAsPqYkTQ/aoFiuqr9hi12xsdGwAAoF8YyQEAAAAAwBpRLTzUK1AE2a/V6I8g24MWOhZPLypzOiNJSu9JK7WQqrvf/IuBDvcDzj3buu25Z4Mdq95xGrXt1jlbaXSeXlxTs3b9Pl+vztlpPJsdr9Ux2o1pp89Yv+5Ft5/Rdu5FJ89NvWN063sLoDcocgAAAAAAgEAaFUTamaLq+OHjOnPijGITMcmQdLOZXbC14/EdOvzq4W52uan5F8MlaYO07aVW/ZLCXVezawpbbOokhmH6GeacncaznfOH2a+buh3TRu06iWkncenWd3YQ9wZAeBQ5AAAAAADAD6hXuKhX2Aha4Dh/6ryO7j+qxJaEtu3bpvhkXIZpyLVdWTlLpeWSzr5+Vi+MvaAjbx5Rem9aUutEaJCkZbMkbJDEZ7tJ3KD96kSruARJPq/eHjQe1W3tJoEbFRZ61c9656w9bqt92z1fWN2KaSthYxq2XacxDRuXRuemcAGsXazJAQAAAAAAbtHu4uGGYay8qn+vdv7Ueb382MtKLaR01yN3ae6hOaX3pJXcldTs/bPafM9mTd81rZntM0psTejYo8d04dsXunpNtYJO6dNov0FOXxMkMdzuMYIUDMIm1dtJgLfarxf3rRvxDHr+2vWbIUwAACAASURBVGMP4jlqN6Zh2nUS07Bx6dZ3NszoHQCDRZEDAAAAANap2sR0mP16sW31PmhPoyJDO/u1W+BYvRD56rU+Vju6/6g2371ZqYWU0nvSuuPhO5R6MKXZj89q092bND0/rQ1zGzSZnNRUakqx22J6ad9LgfuAYIImgfv9i/fa8w1rP4MaxuR42JiO+r0IaxjvIYDGmK4KAAAAANahanK7Ot1Qo8R2s/1abWu0ZkOzbbXnRXCr70P176rVMW613+p/rt7eziLlqx0/fFyJ2YRmts9oy31blFxIanp+WhWrovylvKLjURmGIddxVbEqckrO/8/evQbXcd53nv91nwtAHoAECVIADiTQUiRKNkOLoinFsaWIsT2jJI7jCkVVdMmuHCq2N3FS2omT2a31ahxNade1U5VKaTPOZRxLmR1ZisRQyWTLsWzFkZXRMI4sWuOhlo5lKzJl8gC8Azjn9OnT133ROBB0hHO/A99PVRcb3f308/TTzTfP/zzPX67lyl609fSdT+vAnx9opjta0o5fcreSNBnN6fUv8HsdHFrP31i73v1aC9YA6wVBDgAAAABYpyoNXjdyXb33WK1MJStzPdSSP5PX6W+d1hsvvCFJmrllRtP7ppWaSNXVlrWmUt9Wy61Rz/F6z692zYkjJzRzy4xSEymNTo9qbMeYNs9slj1vy3d8uZYrJ+soOZKMtlRSiVRCw1uGdfyp4x0LcjQymNmOweRuDcAzSPumdr63eq/rZaChH9pQj258o828+0HpPwBvR5ADAAAAANA36l0qqZgt6ks/9yVlvpWRGTMVS8QUhqFe/L9fVBAEmr5pWr/8zC8rsTHRhVajktljs0psTCg5klRiY0JmwpTv+LLnbXm2pzBYmlUSM2TGTZkJU2bSVDwZj7bhuOZentPkDZMttaPaoGq1Ac1K5Rpd+78XgYd+GKg9eX/07KslmO5knzT73qppNsF9p/TDN9aqTvRXq+++H/7fAGgcOTkAAAAAAG23ckmkWnk3Ki1jVcn3/up7+ndb/52yp7Kaef+MrvrQVXrHT79DM++fUXpfWtt2btP5757X5zZ9Tt//8vfb9Uh9bXZ29i1breNHjx5d3n/++ec7tp85lpEMyTANhUEoN+8qfzavbCYr67wlJ+fIsz0FXqAwCKVwaaksUzJiUZnMS5mm+6VdSomQ60mIXCuxdTcG9/txoLYU7Cht3dDIe1tNvf3Z7QBDL7+xVnXrG2303fdznwGojSAHAAAAAKAjypNRl6uUi2NlYKQ8QPLdp7+rvzr0V7r8Jy/XO376HZr+iWml96U1cf2Etr1rm7Zes1VjV45p69VbNTYzpqfveVqv/r+vduT5+sWDDz6o5557btX+Xi0puCQFQfCWazq5r1AK3EBuzlXhYkG5uZyymazyZ/IqXCiouFiUm3fl2Z58148CHn4YBT3apHzAc+XAZ63BzWoDyo0MjHZrmap+C3BUGmRuNvDQSL2VjtXz3hoNcPRDv/dDG6rpZoCj0rHV3n0/vUMAzWG5KgAAAABYp+rNfVHtunruUSmYUStPRPk1vuvr8MHDuuL9V2jy+klte+c2jU6PKhaPyck7KlwoKH82r1gyJsM0osF1P9ATv/CE9LtVH3Hgvfrqq7r77rvfdjydTq96/c0337y8v3///o7tp/dF9Ts5R4X5gobODckwDDlZR0YsSjbu5l0VF4ty8o5c663BDsM0lL5x9Wdoh2rLKQ2SQRik7ee2lWumP6sFTgbh/XTaIPQB7xAYXAQ5AAAAAGAdWrmcVOnvkpXBhWrX1brHyutWWm2WRj3LVD350Sc1evmotly1Rduu26apG6Y09o4xubar/JkouBGGoQIvkO/48mxPruWqOFqU/YQt3Vlf36B9pvZOybVc2fO2rPOW4kNxBV6g5EhSZtxUGITybE9O3lFxoSgntxTosDx5drRN7mktH8daN8iDr/3Y9n5s06CjTwF0GkEOAAAAAFinKgUWas2waOYe9Z6rdF3gBfrBV3+gmZtnlJpIaWRyRKOXj2pkaiRKYl3w5OScKMF1KqHExqUtldDQliHZ37NlqPqMFXTGrjt26bVnX4sCG7Eo8XhyJCkzYUphNEPHtVw5uSjQUZrVYS/Y2n3X7p60uTTDo506seZ/o4PHtWau9MtgdLPtbPW9NfP89SSu71Z/Vnv2Vvu02W+mm0tUNfvu++kdAmgOOTkAAAAAAH1v9tis4sNxJVNJJTYkZMQMeQVPhQsFuZar0A+XE1ybMVNmwlQsEVMsGW1GwlB4un05HlC/g08dlGu5ys5ml/NxZE9nlc1Ef+fP5GWds1S4UFDhYkHFhaLsBVsKpANfOtDRtjWSm2G1Y+UDn5Xut/J4uwZLWx18LW9rpxIv1+qTRpN619vORnMvDMpgdju+sXb1aa1yvepT8m4A6w8zOQAAAABgHRkZGdHOnTv1xhtvLB87deqU3ve+90mSvvzlL+vDH/5w3+3Pvjwb/UzP1HIeh/zZvPyirzAM5eQceQVPgRtECatDSYbeDHyYpsJZghy9cuiFQ3rstseUPZWVa7lKpBKKJ+OSKYV+KN/15Vne8gyOMAx139H72lZ/s8nFS+UaCQB0KlhQqY5av95f7Vi156r1i/xax6sFEBrRbDubeW+t9Gcrmu3TVmYsdKJPa73zRvu02X5p9v8sgMHHTA4AAAAAWEc+/elP673vfa82bNiwvG3dunX5/MTERF/ux4fiUiD5RV9OzpF1wVqeFZA/k1fhQuHN5NW2K9/xo4CHHyoMQoVhqDBOkKNX0jemdc8z9yh3Pqf5H85HszkyWeVmc8uzOfLn8srOZuXmXN377L2a3Nv5XBwn768+cF1t0LdSAKHZurqp2nN1s65a9TXbzlbq7HetfmOd6NN+spbfPYDKmMkBAAAAAOvM9u3bK/69b9++vtxP70svz9goXCooeTYpSXJGHRkxI5rdYblR8ursUvLqorcc7AjCQMY0OTl6afqmaT1QfEBH7jqiV558RfHheDTrRtFsG8/2tPuu3XUvUVXPgGU7BjUbvUc3BlIH7bl6UbZdeTVa0WzgoNX7dqp8t/q0V8/X6XsB6ByCHAAAAACAvnfZ7svkF33Zl2wVzhcUH4or9EMVF4uKJWIKg1CeHS13VAp0OHlHbsGVV/AkXwq3M5OjH9z+xO26/YnbNffynDIvZSRFMz0m93R+5gYAAFh7CHIAAAAAAAbCnnv36MRfnFByNCkjZsh3fCVSCcUSMUmS7/pyLVdOLgp0FBeLcnKO7Kwt3dDjxuNtJm+Y1OQNBDYAAEBrCHIAAAAAAAbCLzzyCzpx5IQWM4sKw2jmRiKVUCwZk2FES1Z5thctW5UtRoGOS0XFh+LyPuJFyciBPtRIkmSWzwEA4K1IPA4AAAAAGBiffPmTCt1Qiz9aXE5enc1klZ3NRgmsz+ZlnbdUuFBQ/nxeZsLUJ1/+ZK+bDQAAgA5hJgcAAAAAYGBsuWqL7vvmffrCTV/QwhsLSqaSim+MyzRNhWG4nIDctVwlU0l94qVPaNPMpl43G6iK2RkAADSPmRwAAAAAgIGy5aot+tfn/7X2fmKvnIITzeSYyyp3JqfcXE6e4+nGT92o3z772wQ4AAAA1jhmcgAAAAAABtJtv3ebbvu925SbzSnzUkaSlL4xrZHJkR63DAAAAN1CkAMAAAAAMNBGpka08yM7e90MAAAA9ADLVQEAAAAAAAAAgIFEkAMAAAAAAAAAAAwkghwAAAAAAAAAAGAgEeQAAAAAAAAAAAADiSAHAAAAAAAAAAAYSAQ5AAAAAAAAAADAQCLIAQAAAAAAAAAABhJBDgAAAAAAAAAAMJAIcgAAAAAAAAAAgIFEkAMAAAAAAAAAAAwkghwAAAAAAAAAAGAgEeQAAAAAAAAAAAADiSAHAAAAAAAAAAAYSAQ5AAAAAAAAAADAQCLIAQAAAAAAAAAABlK81w0AAAAAAACoxTCM5f0wDBu+rlr5lecaLQsAAHqLmRwAAAAAAKCvlYIMpQDDakGJWteFYVgxQFE6t9o1hmG85VylugEAQG8wkwMAAAAAAPS9UvChVqCh3usqKQU1yu/XiNljs8ocy0iS0vvSmto7tep1Ox5u+NaSpJP31y578v7K5yqVrVamVvlm6mu2/kbr6dRzVSrX7fo6WWerfVrpXrXKN9qnrfx/qFS+E++i3d9os/9n6nkX7Xz3QKcR5AAAAAAAAGiDwwcP68SRE0psTEiGpKX4iGu52nXHLh186mDX2rLj4dUHI6sNBpfONTroWU/ZTmu1bZXKV+rHWnVWU6ut7W5ns3W2833X21fN9mkr2t2n3f6/16h67tXP/9eB1RDkAAAAAAAA0NtncdR7/tTRU3r01keV2pbSzC0zSo4kZZiGAjeQk3Nkz9t67dnX9NDQQzr0wiGlb0xLqj2gWc8gYrXB1EqDrbXqXa1crV9+Vytbz0BxOwZMm3mulefL71GrXHmZRgeiK/Vxp9q5Wp3l963n+kbrbEa7+rSWZvu02XKt9mez/dKMXr17oBnk5AAAAAAAAKihWoDj8Y88rqm9U7ryQ1dq+qZppfelNblnUtvfuV1br9mqsSvHNL5zXKmJlB677TGdfvF0R9tazyBpo+dqXdPrwc5Wn2u16+oJGDQ7qN7IAHit6+pdiqnRd9eOPq2n/vL79uJbarRPmynXSn+2q1/qfRftevdAtxDkAAAAAAAAfa+UX6NWno16r2u07kozPB699VFtvXqrpvZOKb0vrcvfe7mm3jOl7e/ari1Xb9HYjjFtmt6kkckRjU6NKrEhoUdueaRtbRtkvViaqFIbag30d7ut5fX1azvr1Y8D4832Ke8C6D8sVwUAAAAAAPpaKYl4KXCxMuCwMgBR67ry/Ur3WWm1oEnpusMHDyu1PaXxnePadt02Te6d1NiOMfmOr/zZvOLDcRmGocAL5Du+PNuTa7myF209fefTOvDnB1rrmDWCQdfO6XX+hF4Hh/i23tSvQRegHZjJAQAAAAAA+l4Yhstb+fF6ryvfqt2nnnInjpzQlqu3KDWR0uj0qMZ2jGnzzGZt3LZRw2PDGto0pORI8s0tlVQildDwlmEdf+p4K91RVScSYtf6dXqvB9OrYXD3TTsefuvWyn3qvaaX30Srz9ktnW5jO9/FIPQn1h9mcgAAAAAAADRo9tisEhsTSo4kldiYkJkw5Tu+7Hlbnu0pDJZmlcQMmXFTZsKUmTQVT8ajbTiuuZfnNHnDZEvtaCZpdC3VluGpNmjcqTwN7dIPAZiVfVgtuXu7dSo41Upuh3brZn92Sif7q9337of/T0AJMzkAAAAAAEDfmp2dfcu28vjZs2f1/PPPLx/r5v43nviGZEiGaSgMQrl5V/mzeWUzWVnnLTk5R57tKfAChUEohUtLXpmSEYvKZF7KNNMlHdEPv7rvhH5+rnbNqmhEKYF1K4ms6+nTbgcYaiVy7+eAR6e/0XY+ez//f8L6xkwOAAAAAADQlx588EFdc8012r9//9vOrczDUX6sG/vRASlwA7k5V4WLBcWH43ItV4ZhyLM9FReLcvOuPNuT7/pRwMMPo6BHm1QabKw0W6DStdXuVc91jdTXLf06IFtp6a9OJ6yuFAho97fST/1eer5+1a0ARzuXqeqH9wqUI8gBAAAAAFhXVksg3ch1jZQvP19vWbzp1Vdf1d133/224+l0WpJ02WWXLR9bGQzp+P7d+/X6H70uJ+eoMF/Q0LkhGYYhJ+vIiEXJxt28q+JiUU7ekWu9NdhhmIbSN6ZrPn+z6h28bkeAo5H6GqmzFYMwINvPbVtNo31aLbgwCO+n07rZB62+C94X+h1BDgAAAADAulEKMoThUr6EVQIRta4r/bsyYFGpfPmx8mAJgY7BNbV3Sq7lyp63ZZ23FB+KK/ACJUeSMuOmwiCUZ3ty8o6KC0U5uaVAh+XJs6Ntck9r+ThatVYHLgf5ufq17f3arkE1SP05SG3F+kWQAwAAAACwrqwMVlQLVNR7XblS8KKRMqs5850zeuO/vKGTL5yUJO24ZYdmbp7RxPUTLd0X7bPrjl167dnXosBGLEo8nhxJykyYUij5ri/XcuXkokBHaVaHvWBr9127e9r2Xg1cdnrpoEafq9YMlH4Z4G22ne1YrqmZPm3XvVpV7dlb7dNmv5lu9kGr76Jfvn+gFhKPAwAAAADQJu2YnXH+e+f1+Xd+Xl+8+Yv62//1b/X6372u1//2dT37O8/qizd/UZ9/5+d16QeX2tRitOLgUwflWq6ys1nl5nLKZrLKns4qm4n+zp/JyzpnqXChoMLFgooLRdkLthRIB750oKNt69RSQZXu2+hAeicGTVsdkC1/hk4FZGr1Yb3LfdW6Xz319tOAfLPq+Sa71ae1yg1Cf5YMUlsBZnIAAAAAANAGtQIc9czu+PZ/+La+8j9/RWM7xjTz/hklR5IyDEO+46uYLcqet5Wby+nzuz+vn//Dn9eeX9nT7sdAgw69cEiP3faYsqeyci1XiVRC8WRcMqXQD+W7vjzLW57BEYah7jt6X9vqrzWoWj5AufL6Wr9yL/+7dH0j5bql2ecqHav2bLUCALWOV/u1fyOabWe9765cK33arGb7tNmAVKf6tNY7b7Q/W/nWmtGLdw+0giAHAAAAAABtUh7EKA98VEpgLkkv/vsX9dwDz+mKn7xCW6/eqpHJEcU3RHkenJyjwsWCrPOWEhsTSp5J6iv3f0W+43f2gVBT+sa07nnmHj1yyyOyF20NbxlWPBmXETMUBqECL5Bne8szOO47ep8m93Y+F0cnBh9L92xkMHilfv5leKVlnTrVj83W1WzZbj5ft1Vbkos+BdYHghwAAAAAgHWl3pwZjebWKJ/FUW1mR/m5/Lm8vvbpr+mK912hiXdPaPu7tmt0elRmzJSTc2RdsJRMJRVLxqKyS4Pnf/ObfyP9lqThupqIDpm+aVoPFB/QkbuO6JUnX1F8OK4wWEpQbxrybE+779pd9xJV9Q7MNqMdA7C9rLuT9270Hq3U2Yuy3Xy+Zu/Zq2+rG/X26ntp5p4EajBoCHIAAAAAANaNUtCiFLgon1lRnmy80nXl+/Xk4VhZrvz6I790RKOXj2rsyjGNXzuuiT0T2nLlFrmWq9xcTmbCVBgsLX1ke3ILrlzLVfJSUvZhW/ofGu0JdMLtT9yu25+4XXMvzynzUkZSNNNjck/nZ24AALBeEeQAAAAAAKwrlQIS5cfrva7eeiqV8wqefvj3P9SOm3cotT2l1GUpjU6OauO2jbLnbTl5R4lsQolUQomNb92GNg/Jft2WWLWqr0zeMKnJGwhsAADQDQQ5AAAAAADoocyxjOLD8Shh9XBchmHIyTuyzlnyip4CN5DCaCaIGTNlxk3FErFoS8ZkxA0p0+unANqrkUTSLK0DAOub2esGAAAAAADQaZOTk7r55pv12muvLW8lp06d0pNPPrn8d7f3z3znjEzDlAzJd305OUf5s3ktZhZlnbdUXCzKK3jyHV+BH0iBpFCSEeV7ME1TmmuiUwAAANYAZnIAAAAAANa8T37yk5qfn1/13ObNm3X99dcv/93tfeclR0EQyC/6y0nGY8mYnLwjwzDkF30Vs0W5eVdewZPnePI9X6EfKgzCaBksEo9jjWF2BgCgXgQ5AAAAAADrwtjY2KrHR0dHdd111y3/3e398+F5hWGoYrYo+5Kt/Nm8JCmZTcqMmwq8QK7lqrhYlJNz5FquPNuTV1ya3REGUrq+PgAAAFhrCHIAAAAAANBD4zvHFYvHZF+yZZ2LZnEEXqDkSFKxRExhEMqzPTl5Jwp0ZB25uaVZHbYnmVK4tb5k6AAAAGsNQQ4AAAAAAHrIiBna+/G9OvaFY0qOJGWYhryitxzkkKJcHZ7lqZgrqrhQVDEbBTuK2aK0T5LR22cAAADoFYIcAAAAAAD02L/8vX+p448f1+KPFhWG0cyNxEgiCnIYUuiF8opetGxVNgp0FC4VtHHrRi3+i8UoETkAAMA6ZPa6AQAAAAAAQPr1V35dRszQwskFZTNZZU9nlc1klZvNKXcmp/zZvKxzlgoXCsqfyyuRSujXXvm1XjcbAACgp5jJAQAAAABAH9gwvkGf+qdP6Qs3fkHZTFaxZEyJVEJGzJBCKfACOTlHgRto0xWb9PFvfVzJkWSvmw0AANBTzOQAAAAAAKBPDG8e1m+++pva/9n9SqQSWnhjQdY5S9Z5S4unFjW0eUgfeOgD+tR3P0WAAwAAQMzkAAAAAACg77z30+/Vez/9XimUMi9lJEnpG9M9bhUAAED/IcgBAAAAAEC/MghuAAAAVMNyVQAAAAAAAAAAYCAR5AAAAAAAAAAAAAOJIAcAAAAAAAAAABhIBDkAAAAAAAAAAMBAIsgBAAAAAAAAAAAGEkEOAAAAAAAAAAAwkAhyAAAAAAAAAACAgUSQAwAAAAAAAAAADCSCHAAAAAAAAAAAYCDFe90AAAAAAAAArD+GYSzvh2HY1HWVzq08Xk8dAIDBxUwOAAAAAAAAdFUpCFEKPKwWlKh1nWEYCsNweSsPeKzcAABrFzM5AAAAAAAA0HWl4EN5gKLe6+oNXpSCIbXMHptV5lhGkpTel9bU3qlVr9vxcF3Vvs3J+2uXPXl/7fuU36NWmUp11lNXI1qtZ7XyneiPdtfXqTrb/d5W3q/Rb6bZb6yV8p14F+3+RjtdrtWy6wlBDgAAAAAAAKxbhw8e1okjJ5TYmJAMSUvxENdyteuOXTr41MGutWXHw40Pete6X61z7Rg0bbWeSuXb3R+1ylWrr1rZZstVKtvu91ZvXzXbp61od59WKtdKn3biW+tk2fWIIAcAAAAAAAAGWqXZGtVmcZw6ekqP3vqoUttSmrllRsmRpAzTUOAGcnKO7Hlbrz37mh4aekiHXjik9I1pSbUHQusZfK42CFtrcLd0rt5B0FrtrVVfvZqtp9IMg071x2p1daOdq9VZft9a1zZaX7Pa1ae1NNunzZZrtU9b7ZdmAhetlF1PyMkBAAAAAACAgVXvclQrnTp6So9/5HFN7Z3SlR+6UtM3TSu9L63JPZPa/s7t2nrNVo1dOabxneNKTaT02G2P6fSLpzv0BJF6BsgbHUSuZ8C2HdpRT/l1neiPaoGoTrWzVvCr0ba085lXu3cvlkNqtE+bKddKnzbbL630Z6/exaAiyAEAAAAAAICuK+XXqJaPo9Z1zQQ4JOnRWx/V1qu3amrvlNL70rr8vZdr6j1T2v6u7dpy9RaN7RjTpulNGpkc0ejUqBIbEnrklkcargfV1RsA6Pav2Mvr69d21qsfB8ub7dNBfxfoDJarAgAAAAAAQFeVkoiXAhcrAxUrAxe1rlv5b7X7rHT44GGltqc0vnNc267bpsm9kxrbMSbf8ZU/m1d8OC7DMBR4gXzHl2d7ci1X9qKtp+98Wgf+/EAbewJrWTvznrRSf6/q68fgCtYmghwAAAAAAADoukozMMqP13tdvedPHDmhmVtmlJpIaXR6VGM7xrR5ZrPseVu+48u1XDlZR8mRZLSlkkqkEhreMqzjTx3vWJCj1wPSg17PIGhHEKDRJOK9DDT0QxvqwTc6+AhyAAAAAAAAYF2YPTarxMaEkiNJJTYmZCZM+Y4ve96WZ3sKg6WZIzFDZtyUmTBlJk3Fk/FoG45r7uU5Td4w2VI7qg2qdntAuFv19cNA98n7o75fLcF0Jwe6K927lSBAswnuO6Wb/dkp/fCNojnk5AAAAAAAAEDXzM7OvmWrdfzEiRPL+88//3xL+5ljGcmQDNNQGIRy867yZ/PKZrKyzltyco4821PgBQqDUAqXlsMyJSMWlcm8lGlfZ/RIt35h38+/5C8FO0pbN5SSSTebVLre/ux2gKFW8vR+Dnj08zeK+jGTAwAAAAAAAF3x4IMP6pprrtH+/fvfdq6eZanash9KgRvIzbkqXCwoPhyXa7kyDEOe7am4WJSbd+XZnnzXjwIefhgFPdqk0oBqpVkG7bTeAxyVElN3OmF1pUBAve+80QBHP/R76fn6VT/1FVpDkAMAAAAAAABd8+qrr+ruu+9+2/F0Or3q9bt27VreXxkcaWZ/dlM0Q8TJOSrMFzR0bkiGYcjJOjJiUbJxN++quFiUk3fkWm8NdhimofSNq7ezHRoZ9G7Geg9wrNTPbSvXTH9WCy4MwvvpNPpgbSHIAQAAAAAAgHVhau+UXMuVPW/LOm8pPhRX4AVKjiRlxk2FQSjP9uTkHRUXinJyS4EOy5NnR9vkntbycfQKAY7a+rHt/dimQUefrj3k5AAAAAAAAMC6seuOXVEOjnOWcnM5ZTPZaJvNKjeXU/5cXtZ5S4VLBRUXisuzOuwFW7vv2t3r5jelXwMctZaI6pfB6Gbb2Wq7m3n+8rwfq+UAaTYnSKOqzSZptU+b/Wb65ZtCexHkAAAAAAAAwLpx8KmDci13OaiRzWSVPR0FOnJzOeXP5GWds1S4UFDhYhTosBdsKZAOfOlAR9vWifwF/RrgqFS+0t/t0mpApdl2rnZdtToHZTC+Vn9K3evTWuUGpU/ROJarAgAAAAAAwLpy6IVDeuy2x5Q9lZVruUqkEoon45IphX4o3/XlWd7yDI4wDHXf0fvaVn+twdhqg961jq8su/JcPb+qb1Yr9axMTr1a2Vq/yK91vJG+rKbZdtYqt5puvbdq9VY7Xl5vswGpTvVprXfeaJ+2u19a+UbrKbseMZMDAAAAAAAA60r6xrTueeYe5c7nNP/D+eUlq3KzueXZHPlzeWVns3Jzru599l5N7u18Lo5uLSPUb6oNZnezrlr1NdvOVursd9Xa36s+xfrDTA4AAAAAAACsO9M3TeuB4gM6ctcRvfLkK4oPxxUGoSTJMA15tqfdd+2ue4mqegZXWxmAbaZstwZ821FPo/fodl+2WrbRvBqd0GzgoNX7dqp8t/q0F/1CsKYxBDkAAAAAAACw/oE9ygAAIABJREFUbt3+xO26/YnbNffynDIvZSRFMz0m93R+5gYAoHUEOQAAAAAAALDuTd4wqckbCGwAwKAhyAEAAAAAAABgWSOJpFlWB0CvkXgcAAAAAAAAAAAMJGZyAAAAAAAAAFjG7AwAg4QgBwAAAAAAa4RhGMv7YRg2dV2te7RSFgAAoN1YrgoAAAAAgDWgFGAoBRdWBhzqva7WPQzDUBiGy1u95wAAADqFmRwAAAAAAKwRpeBCGIYVgxy1rqt0rhTEWE21c+UCN9B/+7P/ph888wOd+e9nJEkT10/o6p+5Wns+tkdmnN9jAgCA+hHkAAAAAAAAdWl1qaqXv/iynvlXz0iSEhsSSo4kFQahfvjcD/WDr/5AX/1XX9XP/fuf0/X3Xt+B1gMAgLWIIAcAAAAAAKjLyuBF+eyNauck6cu/9mUdf/y4JnZPKDWRUnIkKUOGvKKn4mJRhUsFFS4U9OVPfVmz357Vzzz8M51/IAAAMPAIcgAAAAAAgI76q3v/St/76+9p5pYZjV89rpGpEcU3xBW4gYqLRVkXLA2dG1JiOCEzburbX/y2XMuVLu91ywEAQL8jyAEAAAAAwBpRmkFRLR9HrevqvUe9Tv3DKX33yHd1xc1XaGL3hLbv2q5Nl2+SYRpRgOOcpfiG+HIujsAP5Lu+jn/puPTLktJtaQYAAFijCHIAAAAAALAGlAITpeBEpeWjql1Xz7mVf9dz7iu/+RVtntmssR1j2nr1Vk28e0Jbr94qe8FWbjYnSfJdX17Rk2u5cvKOnLyj4kJR7t+40q+23jeDrJ5cJ9WuqzePSvk19dYLAECvmb1uAAAAAAAAaI8wDJe38uP1XNfuc4ULBc0dn9PI1Ig2jG+Iti0blNiYUGI4ofiGuBIbEtE2nHhzfykpueYk2U12xhpQHmyqNLum2nWV3tfKc5W+GYIbAIBBwEwOAAAAAADQEZljGcXiMcU3xBVLxBQGoewFW0bMUOAG8ou+wiCUDMkwDRkxQ2bcjLaEKSNmKMys74H28hk4rV5XyWrJ4gEAGATM5AAAAAAAYIDt2bNHP/uzP6vjx4/r+PHj+oM/+IPlcw8//HBP98//03nF4jEplLyip+JiUfkzeWUzWeXP5VVcKMqxHHlFT4EXKAzCt8xGiBkxGefakxsEnXPptUv62m99TX9yw5/oc5s+p89t+pz+ZO+f6Guf/prmfzjf6+YBANY4ZnIAAAAAADDAPvrRj8pxnOW/Z2Zmlvfvvffenu5vunyTfM+XZ3tyso6s85bMuCkn78gwDflFX07OkZNz5Fqu/KKvwA0U+NHmB760uYHOQFNamcXx7O88q29/8dtSKA2PDWvLO7Yo8ANZZywd+w/H9PIjL2vfr+3TB//PD7a51QAARAhyAAAAAAAw4JLJ5Kr7Y2NjPd1PvyetMAhVXCyqcLGgRCohhZKTdWTGTQV+ECUbzzrRlnfkFlz5tq/ACaKB93QDHYGu+k8f+k+a/fasJvdMamRiRMlU9O25tqviYlH2RVvWeUv/+Af/qDMvn9HdX7m7xy0GAKxFBDkAAAAAAEBHbLpik0YnR2VfsGVtthRLxBS4gZIjSZkJU2EQyi/6ci1XxYWiiovF5VkdTsGRRiVt6vVT9FZplkWtPBv1Xtcuj932mOb++5x27N+h8WvGNTI1ovhwXL7jq7hQlHXBkjVqKTYckxE3dPKFk3r85x+XbuxK8wAA6whBDgAAAAAA0BGGaeh9/8v79OzvPKvESEIyJc/xlExFQQ5JCtxAXsGTk3feEujwLE/6F1Ko9ZsMuxS0KAUuVi4ptXKJqVrXle9Xus9K1cp998h3deofTmnmp2Z02Y9fpondE9p0+SbJkOx5W/mzecWH4zJMQ2EQRknmHV8//MYPo6DVzlZ6BQCAtyLIAQAAAAAAOmbf/7RPL3/xZV34/gWFYSjPjoIcsURMMhTl3liazeHkokCHdd7SxPUTOnXDKa3jGIckVcyVUX683uuavf9Kzz3wnLZcuUWbZzZr649t1fYf367xneMqXCgom8lGM3QcX27Bjd5rPsq7UkwV5X7dJcgBAGgrs9cNAAAAAAAAa9vHX/y4Nl2+SZdev6Ts6Wy0zWaVm8spP5dX/mxe1nlL1gVLubM5bblmi+77h/t63WysIjeb08V/vqiNl23Uhi0bNDQ2tJyLw4ybig3FFB+Ov33bEI9yslyUjHx3ltQCAKwPzOQAAAAAAACdZUi//sqv6/Adh/XPf/vPyp/NK7EhodhQTAqjRNVewZM5ZOq6j16n25+4vdctRgWZY5nlYIYRMxR4gex5O5qV4wXybE+BF0hhtMyVYRoyY6bMeLQZpqEws86n5wAA2oqZHAAAAAAAoCvuOHyH7vzPd+rKD1wpI2Yom4lmdMSSMV31oat091/fTYBD0vPPP69jx469ZSs5duyYvvvd7+rBBx9cPtbN/Sc+/4TM2JtJ44sLReXmcspmsrLOWirOF+XmXXm2J9/1FfqhwiBcDnrEjJh0saluGSilHCn1JIxf7bpq5Veeq3T/eusHgLWAmRwAAAAAAKBrdvzUDu34qR2SoqTjkpaTkCPyjW98Q2EY6nd/93ffdu4973mPJOmzn/3s8rFu7t/1qbv0F3//F/IKnoqLUf4UM2bKyTkyY6Z8x5eTi3JwuNabwY7ACxR4gfzAl7bW3RUDaWWi9lKgoVpy99WuK/1bKUhRLWdKpfoAYK0iyAEAAAAAAHqC4MbgSb8nrcAPVFwsqnCxoMTGhMIgVDKblBk3Ffqh3IIrJ+tEW96RZ3lRsMPxFQahwvTaH4BfGayoNpui3uvq1UyAY/bYrDLHMpKk9L60pvZOrXrdjoeba9PJ+2uXPXl/7fuU36NamXrbWk+9tVSqq957r1a+3f3Rifo6VWer/VntfrXu0Wiftvpdd+tdtPsb7XS5SuWr3YMgBwAAAAAAAOoyMjWi8R8bV+F8QfnNeZlxU77rKzmSVCwRi5axcnw5eUfO4tK2NKvDLbjSNkmpXj/F4FsZECkPalQ7t9Lhg4d14sgJJTYmJEPS0qWu5WrXHbt08KmDbW1zNTsebnzQux9Ua1vpXDODz53oj1ptbXc7m6mz1f5spP5mrmundvdppXKt9GknvrVWy1d6ToIcAAAAAAAAqNsHPvcBHbn7iBKjCRmGIa/oLQc5pGgZMtd25eQcFReLshdtOTlHXsGTfrHHjV8DVgtqrDy2cn+1mR2njp7So7c+qtS2lGZumVFyJCnDNBS4gZycI3ve1mvPvqaHhh7SoRcOKX1jWlLtgdB6Bp+rDcLWGtwtnatnALWeQeJ2zOKodp9az1VphkEn+mO1ct1q52p1lt+31rWN1tesdvVpLc32abPlWu3TVvullVlg5Srdi3mhAAAAAAAAqNu1v3Ct3nHrO7RwckHZ2WyUQP50djmRfO5MTtY5S4ULBdmXbBXni7IuWLrqQ1dJ1/S69evbqaOn9PhHHtfU3ild+aErNX3TtNL70prcM6nt79yurdds1diVYxrfOa7UREqP3faYTr94uqNtqmeAvFOD2q2qZ2C40Xt0oj+qBXY61c5awaRG29LOZ17t3r34xhrt02bKtdKnzfZLK/3ZzHcjEeQAAAAAAABAg+7+8t2auXlGC28sKJfJRQGOTFa52ZxyZ3LKn8krfy4v67yl/Pm8fuyDP6Y7//OdvW5215SWjKqVZ6Pe69rl0Vsf1dart2pq75TS+9K6/L2Xa+o9U9r+ru3acvUWje0Y06bpTRqZHNHo1KgSGxJ65JZHutK2buiXpa/qHcjtdnvL6+vXdtarH4NjzfbpoL+LTmO5KgAAAAAAADTsnq/co69/5ut66Q9fUu5MTrGhmOJD0VCTZ3vyi75iG2J6/++8X/sf3N/bxnZRKYl4KXBRafmoWteV75fOlwdEypeqqpST4/DBw0ptT2l857i2XbdNk3snNbZjTL7jK382r/hwXIZhKPAC+Y4vz/bkWq7sRVtP3/m0Dvz5gdY6po/04+D3WtLuJcGarb9X9fF9NaeV74YgBwAAAAAAAJrywf/jg9r3iX361h9+S69//XVd+P4FSdLWnVt11Qev0k2/cZM2Xb6px63svkoJv8uP13tdveeqnT9x5IRmbplRaiKl0elRje0Y0+aZzbLnbfmOL9dy5WQdJUeS0ZZKKpFKaHjLsI4/dbxjQY61+svztfpczWhHEKDRJOK9DDT0Qxvq0e/faCPfDUEOAAAAAAAANG3zjs360P/1oV43A1XMHptVYmNCyZGkEhsTMhOmfMeXPW/Lsz2FwdKskpghM27KTJgyk6biyXi0Dcc19/KcJm+YbKkd1QZVOz0g3KuB534Y6D55f/T8qyWY7uRAd6V7t/Iumk1w3ynd7M9O6YdvdKVmvhtycgAAAAAAAABtMDs7+5Zt5fFvfvOby38///zzXd3PHMtIhmSYhsIglJt3lT+bVzaTlXXekpNz5NmeAi9QGIRSuLQslikZsahM5qVMU32yHvXzL/lLwY7S1g2lRNTNJqSutz+7HWColTy9nwMe/fyNljTy3RDkAAAAAAAAAFr04IMP6rnnnlMYhstbyWp/d30/lAI3kJtzVbhYUG4uShifP5NX4UJBxcWi3Lwb5VNx/Sjg4YdR0KNNygctVw5e9vOAcCP6dfC40kBxs4GHRuqtdKyed95ogKMf+r0f2lBNP/VVJY1+NyxXBQAAAKAulRKZNnJdrXtUOt9sOQAAuunVV1/V3Xff/bbj6XRa6XR6+e/9+/d3dX92UzSrxMk5KswXNHRuSIZhyMk6MmJRsnE376q4WJSTd+Rabw12GKah9I1vtr/dqi2n1A7dGtQd1MHjftVMf1YLnAzC++m0tdoHBDkAAAAA1FQKIoTh0prdhlE12LDadbXuUe2e1a6rVA4AAESm9k7JtVzZ87as85biQ3EFXqDkSFJm3FQYhPJsT07eUXGhKCe3FOiwPHl2tE3uaS0fx1o3yIPH/dj2fmzToFvLfcpyVQAAAADqUgok1AooVLuu0rlmAxWNlvOLvn70X3+kb/7+N/XN3/+mfnT0R/KLfsP1AgAwaHbdsSvKwXHOWl6qKpvJKjubVW4up/y5vKzzlgqXCiouFJdnddgLtnbftbvXzW9aN5bBanTwuNZyTf0yGN1sO1ttdzPPX2kptJX36PTSXCXVvrlW+7TZb6ZfvqlqWmkbMzkAAAAA9IVKS06VZn6sdq5auXKP//zj+sEzP1B8KP6W4IhX9LTz53bqzr++s+VnAACgXx186qAeGnpI2dmsJMmzPSVTSZlJU4YRLVnl2Z5cK1q2qrhQlL1gS4F04EsHOtq2bgQiOjW42+rgcfkSXZ3qi0pLgTWS86KZdq5Wb7U6B2EwXqrdn1L3+rRWuUHp05Ua/W4IcgAAAADoC5WWoKq1PFWtpatO/v1J/dlP/5k2TW7SzPtnlBxJyjAN+a4vJ+fInrf1xn95Q/82/m/1K3//K7rifVd06hEBAOipQy8c0mO3Pabsqaxcy1UilVA8GZdMKfRD+a4vz/KWZ3CEYaj7jt7XtvprDcbWGjSudrwXA7gr21DPr/fLj5XKrFa21i/yax1vpC+rabadtcqtppX+bEWzfdpsQKpTfVrrnTfap+3ul3q+0Wa+G4nlqgAAAACsYSf//qSe/MUndcVPXKF3fPAdmv6JaaVvTGtyz6S2v3O7xq8Z15Yrt2jrzq3aNL1Jj//84/rRf/1Rr5sNAEBHpG9M655n7lHufE7zP5xfXrIqN5uLlqw6k1f+XF7Z2azcnKt7n71Xk3s7n4ujU8sI9fsv2KsNZnezrlr1NdvOVursd9Xa36s+XSua+W6YyQEAAACgLqVZEiuXh2r0unrv0S7/8QP/UdP7pjWxZ0Lb37ldI+kRxRIxuXlXhYsF5c/mFRuKyYhF7QmCQH/2038mfaYrzQMAoOumb5rWA8UHdOSuI3rlyVcUH44rDJZmT5qGPNvT7rt2171EVT2Dq60MwPaqbLfu3+g9Bq0vG82r0QnNBg5avW+nynerT3vVL83cgyAHAAAAgJpKgYlScKLSElHVrqvn3Mq/Wz335IEnNTIxoq3XbNW267Zp8oZJje0Yk1f0ZJ2zFBuKSZICP5Dv+NE65AVXxfminMOOdLCZngIAYDDc/sTtuv2J2zX38pwyL2UkaXm2IwAMEoIcAAAAAOpSKal3+fFqyb+7ee6f/vKfNHPLjEYmRzSaHtXmmc3adMUm2fO2fMeXk3fk5BwlF5NKppa2jUlt2LJBzv9HkAMAsD5M3jCpyRsIbAAYXAQ5AAAAAKw5mZcySmxMKDmSVHxDXGbClF/0ZV+y5dmewmBpVolpyIybMhPRFkvGFEvGZCQMhbOVAysAgNZVmolX73XVytd7rlbdWF8aSXS8lnIgAIOOxOMAAAAAKorH49q1a5dOnz6t06dP6+jRo8vnvv71r/ft/nOPP7ccxAiDUG7ejRKpZrKyLlhyso4821PgBdE65OHSoJcpGTFDpmHKyHQnbwgArEflSxdWytVU7bowDKvOMqw1C7DWNQCAwcBMDgAAAAAVfeYzn9Hc3Nzy38lkcnl/aGiob/cTyYTCMIyWpco5KlwsKD4Ul5t3JUPyi76Ki0W5eVee7cl3/Sjg4YcKg1ChQn4SBgAdVp7PqdXrOmn22Kwyx5byVuxLa2rvVE/agc5idgYwmAhyAAAAAKhqcnJy1f2bb765b/d/6pd+St//g+/LzbmyL9nKn81LkpycIyNmKHADuZar4mJRTs6Ray0FO5wo2BGEgZRevT8AAIOv3qWyDh88rBNHTiixMSEZkpYudS1Xu+7YpYNPkcAJAHqNIAcAAACANWfyhkl5BU/2gi3rgqX4cFyBF6g4UlQsEVPgB/KL0SyP4mJRTtaRm3flFqJghzwpnGAJEwBYi1bL0VF+7NTRU3r01keV2pbSzC0zSo4kZZhRkNzJObLnbb327Gt6aOghHXrhkNI3EhkHgF4hyAEAAABgTdr9S7v1/b/5vpKppMyYKd/xlUglFEvEJEm+48stuHKyjuwFW8VstHxVcaEovbvHjQcA9Mypo6f0+Ece19TeKY3vHFdqIqXExkSU4ynnqjBfkHXeUnIkqexsVo/d9pjueeaeXjcbANYtghwAAAAA1qQDTxzQQxseUnY2q1ChPNuLghzJKMgReqG8ohcFNrLFKNAxX4yWI/lFLS9JAgDojNIMilp5Nuq9rl0evfVRTe2d0tTeKW27bptGp0dlJky5eVeFiwUNnRtSfCguMxYlb8qeyuqRWx6R/reuNA8AUIZUegAAAADWrI9/8+NyC66yp7PKZt7ccrM55c7klD+bV/58XoWLBRUuFBQaoX71H3+1180GgDWvtDxUKXCxcrmo1fJlVLqudHzlfr3nStvKex4+eFip7SmN7xzXtuu2aXLvpKZvmtZluy7T2DvGNJoeVeqylDZu26gNWzdoeGxYw1uHFU/GpcPt6RsAQGOYyQEAAABgzZq4fkL3fv1e/elP/qmKi0UNbx5WbDgmwzSkUArcQJ7tqZiNZnD86j/+qi7bdZl0pNctB4C1r1LC7/Lj9V7XjnMnjpzQzC0zSk2kNDo9qrEdY9o8s1n2vB0tc2hFyxwmR5LRlkoqkUpoeMuwnBNOxfsCADqHIAcAAACANW3i+gl9xvqM/vJ//Esdf/y4YsmYDEVBDhmS7/p69y+/Wx999KO9bioAoIdmj80qsTGh5EhSiY0JmYkon5M9b8uzPYVBtGSWETNkxk2ZCVNm0lQ8GY9mcsQlY647S2oBAN7EclUAAAAA1oVf/H9+Uf/G+zf6xLc+oQ//8Yf14T/5sD7+0sf1gPsAAQ4A6KJrr71Ws7Ozy5skfec731k+//zzz/dk/xtPfEMyJMM0oiTjeVf5s3llM1lZ5y05OUee7SnwAoVBKIVLy2iZigIfhillGu4OAECLmMkBAAAAYF3Zvmu7tu/a3utmAMC69NnPflaZTKbqklS92o8OREsZurkoyXh8OC7XcmUYRrS84WJRbt6VZ3vyXT8KePhhFPQAAPQEQQ4AAAAAAAB0TTqdftuxPXv2LO/v37+/N/t379frf/S6nJyjwnxBQ+eGZBiGnKwjI2Yo8AK5eVfFxaKcvCPXKgt2hKHCNMEOAOg2ghwAAAAAAABY96b2Tsm1XNnztqzzluJDcQVeoORIUmbcVBiE8mxPTt5RcaEoJ7cU6LC8KGeHF0qTvX4KAFh/CHIAAAAAAAAAknbdsUuvPftaFNiIRYnHkyNJmQlTCiXf9eVarpxcFOgozeqwF2zpx3vdegBYnwhyAAAAAAAAAJIOPnVQDw09pOxsVpLk2Z6SqaTMpCnDiJas8mxPrhUtW1VcKEYBjkDS7ZJYrQoAus7sdQMAAAAAAACAfnHohUNyc66yp7LKZrJazCwql8kpO5tVbi6n/Lm8rHOWChcLsi5aCsNQ9x29r9fNBoB1iyAHAAAAAAAAsCR9Y1r3PHOPcudzmv/hvLKZKNiRm81FQY4zeeXP5ZWdzcrNubr32Xs1uZdkHADQKyxXBQAAAAAAAKwwfdO0Hig+oCN3HdErT76i+HBcYRCtRWWYhjzb0+67duvAlw70uKUAAIIcAAAAAAAAwCpuf+J23f7E7Zp7eU6ZlzKSopkek3uYuQEA/YIgBwAAAAAAAFDF5A2TmryBwAYA9COCHAAAAAAAAEAbGYaxvB+GYUvXGYbxtnO1ytVbPwCsBSQeBwAAAAAAANqkFGAoBRdWBhwava7SsTAMl7fya8rPA8Bax0wOAAAAAAAAoI1KwYXVghD1XlcKVlQrX261WR+1zB6bVebYUr6RfWlN7Z1a9bodDzd022Un769d9uT9te9Tfo96yrRSrl6Vnq3Z9tVbtl390Wx9naqz1f6sdr9a92i0T1v9rrv1Ltr9jXbj/16jz0iQAwAAAAAAAOgjzQQrVpYtqXaPwwcP68SRE0psTEiGpKVLXcvVrjt26eBTB5uqvxk7Hm580Luee3ZatTpK55oZfO52f1Srr1rZZstVKttqfzZSfzPXtVO7+7RSuVb6tFf/95rpG4IcAAAAAAAAQJ+oFeCod3ZIpXudOnpKj976qFLbUpq5ZUbJkaQM01DgBnJyjux5W689+5oeGnpIh144pPSNaUm1B0LrGXyuNghba3C3dK7RAdRmyzV6/3K1nqvSDINu9Ue32rlaneX3rXVto/U1q119WkuzfdpsuVb7tJv/95p9RnJyAAAAAACAvmcYxvLW7HXdPgc0q/y7Kv++ms25ceroKT3+kcc1tXdKV37oSk3fNK30vrQm90xq+zu3a+s1WzV25ZjGd44rNZHSY7c9ptMvnm7bc62mngHyZga1my3XaB3NnKt2XSf6o1ogqlPtrBX8arQt7Xzm1e7d6W+lUr3V/m5HuVb6tJf/9xrtG4IcAAAAAACgr7UjkXOtc5USOTd7DutbpeBEPdet/KZW5uyoVL6RQMejtz6qrVdv1dTeKaX3pXX5ey/X1HumtP1d27Xl6i0a2zGmTdObNDI5otGpUSU2JPTILY/UfX/Up94AQLeXUSqvr1/bWa9eBC5qabZPB/1d1KOVZ2S5KgAAAAAA0Pfakci53nt02oVXL0iSxneO96wN6JzS91UeWJPeGpSodl011XJulH/bK88fPnhYqe0pje8c17brtmly76TGdozJd3zlz+YVH47LMAwFXiDf8eXZnlzLlb1o6+k7n9aBPz/QYE9gvWomf0Yn6u9Vff0YXFnrCHIAAAAAAIB1rdrAcLu8/IWXdfT3j+riDy4qnoyGYzzH0/jV43rfb79Pew7taXud6J1K39BqQYlG71WrTKXzJ46c0MwtM0pNpDQ6PaqxHWPaPLNZ9rwt3/HlWq6crKPkSDLaUkklUgkNbxnW8aeOdyzIMci/PK9mrT5XM9oRBGg0iXgvAw390IZ6rKVvlCAHAAAAAABY18qX/Fnt1/arqWdGiFf09Kc3/akWTi1ow5YNmt43LTNpLid5zp/L66u/9VW9+PkX9fEXPy4jxpJXaL/ZY7NKbEwoOZJUYmNCZsKU7/iy5215tqcwWJpVEjNkxk2ZCVNm0lQ8GY+24bjmXp7T5A2TLbWj2qBqvw8IN6sfnuvk/VHfr5a0uZMD3ZXu3UoQoNkE953Szf7slH74RltFkAMAAAAAAKCK8gBIveeK2aI+f+3nFUvGtOPmHRqZHFEilVAYhPIKnux5W6ntKeXP53Xp9Uv6/Znf12+8+hudfRh01LXXXqvZ2dnlv6empiTpLceaPf7888/r1ltvlaSG97/xxDckQzJMQ2EQys27yp/Ny3d8hUEoJ+fIsz0FXqAwCKVw6Xs2JSMWlcm8lGk5yLFe9PMv+XsxCN9qIKDe/uz2s1VKnl5qx2pBpX7Rz99oMwhyAAAAAACAvleaXVFPIudK19V7j1r3rvfcH/34HymRSujy916u8WvHNZoeVSwZk2dHAQ7rvBX9qj5pyjAMLfxoQX+8+4+le5tqHnrss5/9rDKZzKrfSL3LV1U7vvJYo/vRASlwA7k5V4WLBcWH43ItV4ZhyLM9FReLcvOuPNuT7/pRwMMPo6BHm1QaUK00y2AQ9evgcaWkzZ1OWF0tEFDPO280wNEP/b4y0NGP+qmv2oUgBwAAAAAA6GvtSORcz7mVf6+8/2rHa537u//971RcLOqqD12l7bu2a/L6SW26YpPCIFThQkG5szmZcTMaeC4lei56Wji5IH1D0q0NdRH6RDqd7tjx/fv3N79/9369/kevy8k5KswXNHRuSIZhyMk6MmJRsnE376q4WJSTd+Rabw12GKah9I2rt7UdGhn07meDMHjcz20r10x/VgsuDML76bS12gcEOQAAAAAAQN9rRyLnZs41UybwAr30xy9pfOf4coLn8WvHteWqLSpcLMgwjSioYXly81Gy52K2qORIUkObh+S+6Mr4KXJzoH2m9k7JtdyMScdKAAAgAElEQVTlGUTxobgCL1ByJCkzbkZLqNmenLyj4kJRTm4p0GF58uxom9zDUlXVDPLgcT+2vR/bNOjWcp+avW4AAAAAAADAWnLpny/JszwNbxnW0OiQEhsTMgxDruVGCZ5NI0rsnDAVS8YUG4opPhRXfCiuxIaE5ErhpfYtEQRI0q47dimbyco6Zyk3l1M2k4222axycznlz+VlnbdUuFRQcaG4PKvDXrC1+67dvW5+X2t08LjWElH9MhjdbDtbbXczz3/y/spb+TWdVm02Sat92uw30y/fVDWtPCNBDgAAAAAA0LfGx8eVzWaXt1OnTi2fO3nyZF/uzx6blQwplohJkryiJ+uCpWwmq8KFgty8u5zwOQzD5YTQRsyQGTdlGqaUqdk1QEMOPnVQruUuBzWymayyp6NAR24up/yZvKxzlgoXCipcjAId9oItBdKBLx3oaNv6OX9BLa0OHpc/e6f6otWASrPtXO26anUOwmC8VLs/pe71aa1yg9KnKzX6jCxXBQAAAAAA+tLHPvYxLSwsaHZ2dvnYd77zHd1xxx2SpOeee04f+9jH+m7fOm/JMJfyHBRc2Zds5eZyy0me3UK0RJVX8OQ7vgI3UOAHUhAtgWUapgIraFc3AssOvXBIj932mLKnsnItV4lUQvFkXDKl0A/lu9EyaqUZHGEY6r6j97Wt/loDldUGvWsdLy/bbLlGrLxXPb/eLz9WKrNa2Vq/yK91vJG+rKbZdtYqt5pW+rMV7f7GaulUn9Z65432aS/+7zXbNwQ5AAAAAABAX9qxY8fbju3cuXN5vxRc6Lf9iXdPKAzCKL/BfFH5c3kZpiEn58iMmfIdPzq3WJSbX0rw7PjyXV+hF8oPfIUTLFeF9kvfmNY9z9yjR255RPaireEtw4on4zJihsIgVOAF8mxveQbHfUfv0+TezufiGKRfmLfTygHd8uP9VFezZbv5fN1W6dlK55otv577tKSZZyTIAQAAAAAA0EZT75mS7/gqLhRlXbAU3xBXGIRKZpeSPPtRkudidinvQSnJs+3J+//Zu/8YOc47v/OfqunuGbGH5JBDmt098tD06odtmmdxTOoc7HLFrIXo/lgFWHKEmBIQARQuwAI58O6P/HEJBJmANoe7/CVcNos9ICJwoSVbXAp72DtEAc+xmdMxiC1agCUwOAVcmbuj7qFI0UP2j6muqu66P3p6NBr1j/rV3dUz7xfQcLO6nnqe/la1sPv9zvM8dVdew5Mxx8bjGIy5J+f0cv1lXT5zWR/+5EOlplrPp9RaNs21XB05c8T3ElV+E7phhW07jKRvHH0EvcYoYhmlbdB9NQbBT+FgENcdVPthxXSUv72g16DIAQAAAAAAEKPMdEZf/Ttf1Wf/5TNN7pyUmTLVcBrKZDMy06bkSQ27IafmtAod9+uyy7bsqi27YkvzkpdiJgcG6/Sbp3X6zdNafn9Zxfdam8AUjheUe2LwMzcAIE4UOQAAAAAAAGL2Rxf/SP/ysX+pyqcVyWhtPp7JZjSRaW1G3l4WqL1sVbvQ4VqudHrEg8e2kjuaU+4ohQ0A44siBwAAAAAAQMx2z+/W77/y+3r3n78rr+nJWXU+L3IYa5s8243WJuSVtUJHua4/+JM/0JXqFYmJHMDQBdlIeivtgQCMO4ocAAAAAAAAA3Difzyh9FRa//6f/XvZZVuTOyc1MTkhwzTkeZ6aztpsjrItx3L09/6Xv6fj//i4rpy/MuqhAwAwNihyAAAAAAAADMj3/ofvaf5353XpH1xSZbmihtvQRLq1ZFXDbmgiM6Hsvqz+4f/9D5VbYMkgYJSYnQGMJ4ocAAAAAAAAA1R4sqBzf31Of/P//o0+/unH+ttrfytJ+urvfVWH/u4hzf/e/IhHCADA+KLIAQAAAAAAMGiGNP978xQ0AACImTnqAQAAAAAAAAAAAIRBkQMAAAAAAAAAAIwlihwAAAAAAAAAAGAsUeQAAAAAAAAAAABjiSIHAAAAAAAAAAAYSxQ5AAAAAAAAAADAWKLIAQAAAAAAAAAAxhJFDgAAAAAAAAAAMJYocgAAAAAAAAAAgLFEkQMAAAAAAAAAAIwlihwAAAAAAAAAAGAsUeQAAAAAAAAAAABjiSIHAAAAAAAAAAAYSxQ5AAAAAAAAAADAWKLIAQAAAAAAAAAAxhJFDgAAAAAAAAAAMJZSox4AAAAAAAAAth/DMNbfe54X+Lx+7f2089M/ACDZmMkBAAAAAACAoWoXGtrFhU6Fh37neZ7XtThhGMb6557ndWzXqz0AYHwwkwMAAAAAAABD1y4wbC5ChD2vU5t+2sWQfkrXSypeL0qSCscKyi/kO5538DVf3X7JrXP92946F+yaG6/Xr22nvoP2F/T6QfoIO77N7Ybd36D6jBrPXtcL+qyEebaith/EvYj7GQ1zL4Lch059hu13K6DIAQAAAAAAgC3H73JYvVxavKQbl28ovSMtGZLWLuPUHB1+7rAW31qMYaT+HHwtWsI86HlB+ovShxQu+dxrfGGLTVHiEWacYfqMGs8g/Yc5L05xx7RbuygxjSsuQa4T9zOwFVDkAAAAAAAAwJazeR+OzYWOXrM4lq4t6cJTF5Tdl9X8iXllpjMyTENNpym7YstasXTzyk29Ovmqzr57VoXjBUn9E6F+Eo+9krBxFR769Rl3f/3i4if5vPFzv+NrfxY0ER00HlHH2anPzdftd27Q/sKKK6b9hI1p2HZRYxo2LkH1m+0xjGcgidiTAwAAAAAAAFizdG1Jbzz7hvILeR16+pDmnpxT4VhBuSdy2v/N/dr76F7NHJrR7GOzyh7I6uIzF/XJLz4Z6JjC/GW+32WAgiR8w/CTGA56DT8Fg7BJ9SjxCDrOfvcq6Fji/M6drj2KpHnQmIZpFyWmccQlzOyLIM/MdkCRAwAAAAAAAEPXXk6q3z4bfs/r1CaMC09d0N5H9iq/kFfhWEEPf+9h5b+b1/5v7deeR/Zo5uCMds3t0nRuWjvzO5V+KK3XT7weur9BGMZ+GoPmtwAw7PFt7i+p4/QriUnxsDEd53uRxPswTliuCgAAAAAAAEPV3kS8XYzotrRUv/M2v++2SbnfpaouLV5Sdn9Ws4/Nat839im3kNPMwRk17Iaqn1aVmkrJMAw13aYadkOu5cqpObIeWHr7B2/r1I9PRYpLVElM3qKzUe+dMOriEEn9Fn6z8WAmBwAAAAAAAIbO87z11+bjfs/b/PLTrlMfbTcu39CeR/YoeyCrnXM7NXNwRrvnd2vHvh2ampnS5K5JZaYzn7+yGaWzaU3tmdIHb30QJgy++EmEjjppHgYJ3s8dfO2Lr7DXCHLeKJ+VKN9zmAY5xijLVPXaXD3oNbcCZnIAAAAAAABg2ytdLym9I63MdEbpHWmZaVMNuyFrxZJrufKaa7NKJgyZKVNm2pSZMZXKpFqvqZSW319W7mgu0jh6JVXDbF7d69x2ornbRtvDkoSE7KjiMYhkddgN7gdl1M9XHAa9kXvQNr2KREn4PQ0bMzkAAAAAAAAwNKVS6Quvtl//+tfr769evTr098XrRcmQDNOQ1/TkVB1VP62qXCyrdrcmu2LLtVw13aa8pid5a8tkmZIx0WpTfK8YPCAxiJo0jmMWQZg+pWQmZEcRj/YG1mE3sg664fyw9Ns8PckFj0E+o0n+3uOImRwAAAAAAAAYivPnz+vRRx/VyZMnv/TZxiWkRvVentR0mnIqjlbvrSo1lZJTc2QYhlzLVf1BXU7VkWu5ajiNVsGj4bWKHjHpllDtN8sgyl/8d9svYVCJ2KQWOEYVj26FgG73fLOgBY4kxL39/ZJqGAWOsNfu1d7vM7PVUOQAAAAAAADA0Hz00Ud6/vnnv3T8O9/5zvr7jUWQYb0vHCtIkuyKrdWVVU3emZRhGLLLtoyJ1mbjTtVR/UFddtWWU/tiscMwDRWOF3p+9yj6Jb17JYz7JVWHmRBNUqK9mySPbbMw8YzyrGwHw4pBmPvg57e8HQsdFDkAAAAAAACw7eUX8nJqjqwVS7W7NaUmU2q6TWWmMzJTprymJ9dyZVdt1e/XZVfWCh01V67VeuWeiLYfR5IMItE7zgn0JI49iWMad8R0PFHkAAAAAAAAACQdfu6wbl652SpsTLQ2Hs9MZ2SmTcmTGk5DTs2RXWkVOtqzOqz7lo6cOTKSMfdKxiYpYRt0LH5nroz6u4UdZ9TlmsJ8/yQ9K72+e9SYhn1mhhWDJN2HrYKNxwEAAAAAAABJi28tyqk5KpfKqixXVC6WVf6krHKx9e/q7apqd2pa/WxVq/dWVb9fl3XfkprSqR+dGujYBrF/Qbdrxp1ojWsPgm7/jkvUeIQdZ6fz+u274Gc8o9YvntLwYtqv3bjEtM1PbLcTZnIAAAAAAAAAa86+e1YXn7mo8lJZTs1ROptWKpOSTMlreGo4Dbk1d30Gh+d5eunaS7H13y9JGXcSdtBJ0Y3X7/fX+52Otdt0atvvL/L7HQ+6t0k3YcfZr10nUeIZRdiYhn2+BhXTfvc8aEyjPGth+H1mxqVYExdmcgAAAAAAAABrCscLeuGdF1S5W9HKb1ZaszmKZVVKlfXZHNU7VZVLZTkVRy9eeVG5hcHvxXHrXPyJy16J4qQkSYe5WXqUeIQd5zjcg7B6jX9UMd0KesVuKzw3YTCTAwAAAAAAANhg7sk5vVx/WZfPXNaHP/lQqamUvKYnSTJMQ67l6siZI76XqPKTdBxm0n7Q/cZ9/aDXiNLnKNrGta9GFGELB1GvO6j2w4rpsAqPw+h7nFHkAAAAAAAAADo4/eZpnX7ztJbfX1bxvaKk1kyP3BODn7kBAPCHIgcAAAAAAADQQ+5oTrmjFDYAIIkocgAAAAAAACSUYRjr7z3PC3Vet8/8tvHTP7aPIBtJs5wOgGFg43EAAAAAAIAEahca2sWFToWHfucZhiHP89Zfmwsb3QoXG9tQ3AAAJBkzOQAAAAAAABKqXWDYXKDwe14cBYp2oaSf0vWSitfX9q04VlB+IR+5byQPszMAJA1FDgAAAAAAAIR2afGSbly+ofSOtGRIWquHODVHh587rMW3Fkc6PgDA1kaRAwAAAAAAYBvwOyPDb5ula0u68NQFZfdlNX9iXpnpjAzTUNNpyq7YslYs3bxyU69Ovqqz755V4Xghjq8BAMAXUOQAAAAAAADY4sIUOHpZurakN559Q/mFvGYfm1X2QFbpHWl5TU9OxdHqyqpqd2vKTGdULpV18ZmLeuGdF2LrHwCANoocAAAAAAAACdUuTvTaj6PfeXEXOCTpwlMXlF/IK7+Q175v7NPOuZ0y06acqqPVe6uavDOp1GRK5oQpSSovlfX6idelfxrrMAAAoMgBAAAAAACQRO2iRbtwsbFQsbFw0e+8jf+78fONx/r1sdGlxUvK7s9q9rFZ7fvGPuUWcpo5OKOG3VD106pSUykZhqGm21TDbsi1XDk1R9YDS/YlW2KLDgBAjChyAAAAAAAAJFS3GRibj/s9z+9nvT6/cfmG5k/MK3sgq51zOzVzcEa753fLWrHUsBtyao7ssq3MdKb1ymaUzqY1tWdK9g27Z58AAARljnoAAAAAAAAAGA+l6yWld6SVmc4ovSMtM22qYTdkrVhyLVdec21WyYQhM2XKTJsyM6ZSmZRSmZSUkozl3ktvAQAQBEUOAAAAAACAhHn88cdVKpXWX20bjy0vL+vq1avrnw3jffF6UTIkwzRam4xXHVU/rapcLKt2tya7Ysu1XDXdprymJ3lrS2GZahU+DFMqRokMAABfxHJVAAAAAAAACfLKK6+oWCx2XC6q154ZQ3vvSU2nKafS2mQ8NZWSU3NkGIZcy1X9QV1O1ZFruWo4jVbBo+G1ih4AAMSMIgcAAAAAAEDCFAoFX8dzudz6+5MnTw78fWlXa1aJXbG1urKqyTuTMgxDdtmWMdHabNypOqo/qMuu2nJqm4odnievQLEDABAfihwAAAAAAADwJb+Ql1NzZK1Yqt2tKTWZUtNtKjOdkZky5TU9uZYru2qrfr8uu7JW6Ki5rT07XE/K9e8HAAC/KHIAAAAAAADAt8PPHdbNKzdbhY2J1sbjmemMzLQpeVLDacipObIrrUJHe1aHdd+Svj3q0QMAthqKHAAAAAAAAPBt8a1FvTr5qsqlsiTJtVxlshmZGVOG0VqyyrVcObXWslX1+/VWgaMp6bQkVqsCAMTIHPUAAAAAAAAAMF7OvntWTsVReamscrGsB8UHqhQrKpfKqixXVL1TVe1OTav3VlW7V5PneXrp2kujHjYAYAuiyAEAAAAAAIBACscLeuGdF1S5W9HKb1ZULraKHZVSpVXkuF1V9U5V5VJZTsXRi1deVG6BzTgAAPFjuSoAAAAAAAAENvfknF6uv6zLZy7rw598qNRUSl6ztRaVYRpyLVdHzhzRqR+dGvFIAQBbGUUOAAAAAAAAhHb6zdM6/eZpLb+/rOJ7RUmtmR65J5i5AQAYPIocAAAAAAAAiCx3NKfcUQobAIDhosgBAAAAAACAoTMMY/2953mRzjMM40ufdWu38bif/gEAycbG4wAAAAAAABiqdqGhXVzoVHjwe16vY53aeZ73hRcAYLwxkwMAAAAAAABD1y4weJ7XtcjR77z2DI5eszN6Xb/TDJBOStdLKl5f22/kWEH5hXzH8w6+1vdSHd0617/trXPdP+vWtlebXu38tvcr7Ph6tffTdnO7Yfc3qD6jxrPX9YI+M4N+xoZ1L+J+RsPGZRj3cCuiyAEAAAAAAICx47dAEcWlxUu6cfmG0jvSkiFprTun5ujwc4e1+NbiQPvf6OBrnZOXvZLI7c9GmfSMOr5u7bvFo1+fvfQba9zjDNNn3Pfbb6zCxjSKuGM6iN9QmLiM6h5uZRQ5AAAAAAAAMFbiKHD0usbStSVdeOqCsvuymj8xr8x0RoZpqOk0ZVdsWSuWbl65qVcnX9XZd8+qcLwgqX8i1E/islcStldyN2w7v+OKol9c/CSfN34e9HsFTQJ3KywMapyd+tx83X7nBu0vrLhi2k/YmIZtFzWmYeIS5TeLL2JPDgAAAAAAAIwdwzDWX+1/x2Hp2pLeePYN5RfyOvT0Ic09OafCsYJyT+S0/5v7tffRvZo5NKPZx2aVPZDVxWcu6pNffBJL3930S64GKXwMWxzFlc3n+SkYhE2qB0mA9zvP75JFQe5fnMWqoMW3UTxPQWMapl2UmIaJS5y/2STM1koCihwAAAAAAAAYOr/FiU7nddo8fPOsjLDFjwtPXdDeR/Yqv5BX4VhBD3/vYeW/m9f+b+3Xnkf2aObgjHbN7dJ0blo78zuVfiit10+8HqgP9Oe3ADDspXo295fUcfqVxOR42JiO+70IK4n3cNhYrgoAAAAAAABD1d4MvF2A2Fig2LiMVK/zwl5/cx8bXVq8pOz+rGYfm9W+b+xTbiGnmYMzatgNVT+tKjWVkmEYarpNNeyGXMuVU3NkPbD09g/e1qkfnwoejAQIu0E3whv1X+CPujjEMxbdVivWREGRAwAAAAAAAEPXrWCx+bifwkanc3q16/bZjcs3NH9iXtkDWe2c26mZgzPaPb9b1oqlht2QU3Nkl21lpjOtVzajdDatqT1T+uCtDwZW5Ii6kXZSl8AhSfu5OIoAQTcRT8KG9EkvdoyqGBR1qbPthiIHAAAAAAAAtr3S9ZLSO9LKTGeU3pGWmTbVsBuyViy5liuvuTY7ZMKQmTJlpk2ZGVOpTKr1mkpp+f1l5Y7mIo2jV1LVb+IzSJtum2wPUxIStbfOtb57pw2fBxmTbteOksQOu8H9oCThGYtqUPEK85sNet52wJ4cAAAAAAAAGJrHH39cpVJp/dW28Zif4ysrK7p69er6v6O+L14vSoZkmIa8pien6qj6aVXlYlm1uzXZFVuu5arpNuU1Pclb2+/DlIyJVpvie8UYIhSvoEWTjceGkdxPYqK2Xexov4ahvRl12A2+g84AGJZRPmNRjeoZ7ReTJMdsVJjJAQAAAAAAgKF45ZVXVCwWAy0v5XfZqVjee1LTacqpOFq9t6rUVEpOzZFhGHItV/UHdTlVR67lquE0WgWPhtcqesSkW0K12yyDfm39tOt0ne1Y4Oi2MfWgN6zuVgjwe+/GcYmjQT9jUQ0rVkF/s0m6h0lCkQMAAAAAAABDUygUYjk+MzOjkydPrv876vvSrtYsEbtia3VlVZN3JmUYhuyyLWOitdm4U3VUf1CXXbXl1L5Y7DBMQ4XjnccahyBJ7zjaDco4JGmTPLbNwsSzV3FhHO7PoI06Bn5+s9zDL6LIAQAAAAAAgG0vv5CXU3NkrViq3a0pNZlS020qM52RmTLlNT25liu7aqt+vy67slboqLlyrdYr90S0/Ti2unFOviZx7Ekc07gjpuOJIgcAAAAAAAAg6fBzh3Xzys1WYWOitfF4ZjojM21KntRwGnJqjuxKq9DRntVh3bd05MyRUQ8/NoNYRiho8rjfX7MnJRkddpxRl2sK8/17nTvsePbbKyZKTMM+M0l5pnpJ0j1MEjYeBwAAAAAAACQtvrUop+aoXCqrslxRuVhW+ZOyysXWv6u3q6rdqWn1s1Wt3ltV/X5d1n1LakqnfnRqoGPrlhQOu0mxn+NxJUujJl83j3VQezn0i0nQTb39jrPTeb36HJdkdhzPWFwx9fs7GUZM2Vg8fszkAAAAAAAAANacffesLj5zUeWlspyao3Q2rVQmJZmS1/DUcBpya+76DA7P8/TStZdi679fgrNX0nsQ7aLa2Ee/v97vdKzdplPboPsVdNtQ3E/bXsKOs1+7TqLEM4qwMQ37jA0qpv3uedCYDjIuSS9iJQkzOQAAAAAAAIA1heMFvfDOC6rcrWjlNyut2RzFsiqlyvpsjuqdqsqlspyKoxevvKjcwuD34rh1rnshIO523dqMQq9k9jD78hPnIMfj6DPpoj5jg4hpEoT9zaI7ZnIAAAAAAAAAG8w9OaeX6y/r8pnL+vAnHyo1lZLX9CRJhmnItVwdOXPE9xJVfhKWUZKaYdsOI5EaRx9BrzGKWEZpG9e+GlGELRxEve6g2g8rpkn67W3nwghFDgAAAAAAAKCD02+e1uk3T2v5/WUV3ytKas30yD0x+JkbAAB/KHIAAAAAAAAAPeSO5pQ7SmEDAJKIIgcAAAAAAAAAX4JsJL2dl88BMDxsPA4AAAAAAAAAAMYSMzkAAAAAAAAA+MLsDABJw0wOAAAAAAAAAAAwlihyAAAAAAAAAACAsUSRAwAAAAAAAAAAjCWKHAAAAAAAAAAAYCxR5AAAAAAAAAAAAGOJIgcAAAAAAAAAABhLFDkAAAAAAAAAAMBYosgBAAAAAAAAAADGEkUOAAAAAAAAAAAwlihyAAAAAAAAAACAsUSRAwAAAAAAAAAAjCWKHAAAAAAAAAAAYCxR5AAAAAAAAAAAAGOJIgcAAAAAAAAAABhLFDkAAAAAAAAAAMBYosgBAAAAAAAAAADGEkUOAAAAAAAAAAAwlihyAAAAAAAAAACAsZQa9QAAAAAAAKNhGMb6e8/zQp3n5xqGYYRqBwAAAPTDTA4AAAAA2IbaRYZ2gWFj0cHvee3iRfvV6RrdjvVrBwAAAPjBTA4AAAAA2KbahYt+hYZu5/WbgdEuZmy+dpiZG6XrJRWvFyVJhWMF5RfyHc87+FrgS0uSbp3r3/bWue6fRWnbqX2/84PqNj6//XRq76dt2O8VV3+D6jNqPHtdL+5nJe5n00+bMO3ifkYHHZdu14j7twsA6I8iBwAAAAAgtG5Fj05LVPlpt9mlxUu6cfmG0jvSkiFp7VSn5ujwc4e1+NZi+MEHdPC14RUfhtVH+7Mwyede8Qj7vfqNNe5xhukzajyD9B/mvDjFHdNu7aLEdBRx6dXvIP47AQDojSIHAAAAACC0ToWNfgWObu02Wrq2pAtPXVB2X1bzJ+aVmc7IMA01nabsii1rxdLNKzf16uSrOvvuWRWOFyT1T4T6ST72SsL2S2CGTW622w0qYdsvLn6Szxs/DxqPoN+rW2FhUOPs1Ofm6/Y7N2h/YcUV037CxjRsu6gxHVZcNvcR9nkDAMSHPTkAAAAAALEzDGP91f63X0vXlvTGs28ov5DXoacPae7JORWOFZR7Iqf939yvvY/u1cyhGc0+NqvsgawuPnNRn/zik0F9FUmDTVgPOhkaR1Fm83l+CgZhk+pBEuD9zvO7ZFG384KOJc7v3Onao0icB41pmHZRYjqquLT77vVvAMBwUOQAAAAAgG3KbwGi23nd2m3cVHzjfh792rVdeOqC9j6yV/mFvArHCnr4ew8r/9289n9rv/Y8skczB2e0a26XpnPT2pnfqfRDab1+4vXeXxaB+S0ADHu5oM39JXWcfiUxMR42puN+L/zYDt8RAMYNy1UBAAAAwDbUXlaqXXDotnxUr/OCbkTup92lxUvK7s9q9rFZ7fvGPuUWcpo5OKOG3VD106pSUykZhqGm21TDbsi1XDk1R9YDS2//4G2d+vGpENGIz6A3EEd8wuyfMYj+R9Ufz2YLcQGA8UeRAwAAAAC2qW5Fic3HexUv/BQ2Op3Trd2Nyzc0f2Je2QNZ7ZzbqZmDM9o9v1vWiqWG3ZBTc2SXbWWmM61XNqN0Nq2pPVP64K0PBlbkiLqRdlITp/y1+efiSHYH3UR8lM9FEsbgx6iKQUmPCwDgcxQ5AAAAAACJULpeUnpHWpnpjNI70jLTphp2Q9aKJddy5TXXZpVMGDJTpsy0KTNjKpVJtV5TKS2/v6zc0VykcfRKqgbZO2CcCghJSOjeOteKWadNmwcZy27XjpLsDrvB/aCM87PZNoh4bYW4AADYkwMAAAAAtp1SqfSFV5Djv/3tb3X16tX1z+J8X7xelAzJMA15TU9O1VH106rKxbJqd2uyK7Zcy1XTbcprepK3tr+HKRkTrTbF94oxRCi4fhtUJzF5muS/WG8XO9qvYRAtCgcAACAASURBVGhvYB12I2u/8Rz2szCOz2bbIJ/RcY4LAOCLKHIAAAAAwDZy/vx5/exnP/vSxuBS5w3DNx9v/3vjZ7G+96Sm05RTcbR6b1WV5YrKxbKqt6ta/WxV9Qd1OVVHruWq4TRaBY+G1yp6xGRzsntj0jto4jOJBQQpuQWObgWGsIWHIP12O+bnngctcCQh7kkYQy+jilXS4wIA+DKWqwIAAACAbeajjz7S888//6XjhUKh4/mbj588eXIg70u7WrNH7Iqt1ZVVTd6ZlGEYssu2jInWZuNO1VH9QV121ZZT+2KxwzANFY53/g5x6LWc0jhJUqK9mySPbbMw8exVOBmH+zNoxAAAEARFDgAAAABAIuQX8nJqjqwVS7W7NaUmU2q6TWWmMzJTprymJ9dyZVdt1e/XZVfWCh01V67VeuWeiLYfx1Y3zsnjJI49iWMad8QUABAUy1UBAAAAABLj8HOHW3tw3KmtL1VVLpZVLpVVWa6oeqeq2t2aVn+7qvr9+vqsDuu+pSNnjox6+F+SpHX9gyaP+y3XlJRkdNhxRh13mO/fbSm0jdcY9NJcbb2ezagxDfvMJOGZ6vebHZffBQBsJxQ5AAAAAACJsfjWopyas17UKBfLKn/SKnRUliuq3q6qdqem1c9WtXqvVeiw7ltSUzr1o1MDHVu/pGav46NOeEZNvG7+joMq3kRNHIcdZ6fzevU5LonsOJ7NuGLar90wYxrXb3ZYvwsAQG8sVwUAAAAASJSz757VxWcuqrxUllNzlM6mlcqkJFPyGp4aTkNuzV2fweF5nl669lJs/fv9S+4gbYL2tfl4lMTvxmv5+ev9zcfabTq17fcX+f2OxxXLsOPs166TKPGMImxMwz6bg4ppv3seNKbDjkv7WmFiAwAYDGZyAAAAAAASpXC8oBfeeUGVuxWt/GZlfcmqSqmyPpujeqeqcqksp+LoxSsvKrcw+L04ui0j1CuhOaylhwapVzJ7mH316y/sOKP0mXRRn81BxDQJ4vjNJv07AsB2wkwOAAAAAEDizD05p5frL+vymcv68CcfKjWVktf0JEmGaci1XB05c8T3ElVRkpZxXT/uPofZR9BrjCKWUdoG3VdjEMIWDqJed1DthxXTUf72KGgAQDJQ5AAAAAAAJNbpN0/r9Juntfz+sorvFSW1Znrknhj8zA0AAAAkH0UOAAAAAEDi5Y7mlDtKYQMAAABfRJEDAAAAAIAxEmTDZJbTAQAAWx0bjwMAAAAAAAAAgLHETA4AAAAAAMYIszMAAAA+x0wOAAAAAAAAAAAwlihyAAAAAAAAAACAsUSRAwAAAAAAAAAAjCWKHAAAAAAAAAAAYCxR5AAAAAAAAAAAAGOJIgcAAAAAAAAAABhLFDkAAAAAAAAAAMBYSo16AAAAAACQZIZhrL/3PC/web3ab/ys3+e9+gYAAAC2K2ZyAAAAAEAX7SJDu8CwuSjh5zzP83oWKNqfdypwbPysW98AAADAdkaRAwAAAAB6aBcf+s2k8HseAAAAgPiwXBUAAAAAjFAcS1L99uZv9cs//aU+/tnHunfzniRp7yN7dejvHtKT/92TmvnaTCxjBQAAAJKGIgcAAAAAjEi3Jaran/lZourKP7miX/3rX0meNDUzpT1f26Nmo6na7Zqu/2/X9f7r7+vYHx/T9//59wfyHQAAAIBRosgBAAAAAAnVbQPztn/z9L9R6Vcl5Z7IafrAtDLZjCTJsRzVH9Rl3bNUu1vTf/pf/5Nuv39bz//b54c2dgAAAGAYKHIAAAAAQA/t2RX9ZlX4PS/KGDa6+MxFLf96WQdPHtTso7Oazk8rNZVSw26ofr+u2mc11XbWNDE1ISNl6Na7t/TGH74hHY99eAAAAMDIUOQAAAAAgC7aRYt24WLzzIrNS0t1O2/z+/bnmwsi3dptLnD858v/WUv/cUnzvz+vr3z7Kzpw5IB2PbxLMiRrxVL106pSUykZpiGv6anpNNWwG/rNz38j7ZL0WNiIAAAAAMlCkQMAAAAAeui2Gfjm437Pi+Ozn738M+05tEe753dr7+/s1f5v79fsY7Na/WxV5WJZXtNTw27IWXXk1BzZVVt2xVY9W5fzU4ciBwAAALYMc9QDAAAAAAD4VylVdO+v72nHV3booT0PaXJmcn0vDjNlamJyQqmp1JdfD6WUzqale5JRjX9JLQAAAGAUmMkBAAAAAGOkeL24XswwJgw13aasFUsypKbblGu5arpNyWsteWWYhswJU2aq9TJMQ16x+ywRAAAAYJwwkwMAAAAANjl58qSeffZZXb9+ff3Vdv36df3lX/7l+r/Pnz8/1Pcrf70ic8JsLUlVb20yXlmuqFwsq/ZpTfWVupyqI9dy1XAa8hqevKa3XvSYMCake6HCMlbae6T42TC+03m92m/8bPM5fvsFAABAPAyv10KvMTh//rw8z9MPf/jDQXaDmG3l+7aVvxswKPxuAABIjo/+z4/0F//gLzT35Jz2fWOfZg7NaPrAtDI7MzInTDXshuyKLWvFUu2zmmp3aqreqap6u6rKckUrf7si7zlPr/zolVF/lYHZuMF7p83gg5y3cYP5Xv2FaQcAAIDoWK4KAAAAAMZI4bsFNRtN1R/UtXpvVekdaXlNT5lyRmbKlNfw5Kw6sst261W15dbc1swOuyGv6ckrbP3ke7vAsLGAEeW8buIoZpSul1S8XpQkFY4VlF/Idzzv4Gvhrn/rXP+2t851/yxK217XCtLO7zU38nv9Tu39tN3cbtj9DarPqPHsdb1+1wga06jP5rDuRdzP6CDjEvf9B4BhoMgBAAAAAGNkOj+t2d+Z1erdVVV3V2WmTDWchjLTGU2kJ1rLWNkN2VVb9oO1V8WWU3PkrDrSPknZUX8LSNKlxUu6cfmG0jvSkiFprVbi1Bwdfu6wFt9aHNpYDr42+CRm2CJN2Gu2PwuT0O0Vj7Dfo99Y4x5nmD6jxjNI/2HOi1PcMe3WLkpMhx2XuO8/AAwLRQ4AAAAAGDN/8D/9gS4/f1npnWkZhiG37q4XOSSp6TTlWI7siq36g7qsB5bsii131ZX+aMSD30LCzuJYurakC09dUHZfVvMn5pWZzsgwDTWd5vpSYzev3NSrk6/q7LtnVThekNQ/Eeon+dgrCdsvuZvU5Ga/uPhJPm/8PGg8giaiuxUWBjXOTn1uvm6/c4P2F1ZcMe0nbEzDtosa02HFpVe7Ydx/AAiLjccBAAAAYMw8/vcf19ee+pru37qvcqmscrGs8idr/1sqq3K7otqdmlY/W5X1W0v1lbpqn9X09ae/Lj066tFvb0vXlvTGs28ov5DXoacPae7JORWOFZR7Iqf939yvvY/u1cyhGc0+NqvsgawuPnNRn/zik4GOaRgJy0H9FXgcRZnN5/kpGIRNqgdJgPc7z++SRd3OCzqWOL9zp2uPInEeNKZh2kWJ6bDj0qs/ChsAkowiBwAAAACMoef/r+c1/3vzuv8391UpVloFjmJZlVJFldsVVW9XVb1TVe1uTdW7Vf3O939HP/g/fjDqYQ9Ne3+Nfvts+D0vLheeuqC9j+xVfiGvwrGCHv7ew8p/N6/939qvPY/s0czBGe2a26Xp3LR25ncq/VBar594fShjG7QkJUn9FgBGvVxQUsfpV5LueVvYmI77vQCArYzlqgAAAABgTL3wb1/QT//ZT/Xev3pPldsVTUxOKDXZ+n/zXMtVo97QxEMT+t1/8rs6ef7kaAc7RO1NxNuFi41LSm1cYqrfeZvfd7vORr3aXVq8pOz+rGYfm9W+b+xTbiGnmYMzatgNVT+tKjWVkmEYarpNNeyGXMuVU3NkPbD09g/e1qkfn4oWmIji3GgbgzXq/RNGXRxKYnFlFIgLgO2CIgcAAAAAjLHv/8n3dewfHdMv/9Uv9fFPP9Zn/+UzSdLex/bq69//up78x09q18O7RjzK4eu2V8bm437PC3v9jW5cvqH5E/PKHshq59xOzRyc0e753bJWLDXshpyaI7tsKzOdab2yGaWzaU3tmdIHb30wsCJH1I20/WzSPIrkKsWVz8WR7A66ifgoE+pJGIMfoyoGRbn/SY8pgO2JIgcAAAAAjLndB3fr6f/56VEPAz2UrpeU3pFWZjqj9I60zLSpht2QtWLJtVx5zbVZJROGzJQpM23KzJhKZVKt11RKy+8vK3c0F2kcvZKqQfYOCJKcHXVSdNT9t8dw8LXOmzYPMtHd7dpREtZhN7gflGHGc1AGEa+ocel0fhJ+SwDQCXtyAAAAAAC2jFKp9IXXxuN3797V1atX148N8/3P3/y5ZEiGachrenKqjqqfVlUullW7W5NdseVarppuU17Tk7y15a5MyZhotSm+VwwTksj6bVDdL5E+Ckn+q/N2saP9Gob2htJhN7L2G89h3/Owz2YSDPIZHVRckhxPANsbMzkAAAAAAFvC+fPn9eijj+rkyZNf+mzjPhybjw3jfeuA1HSaciqOVu+tKjWVklNzZBiGXMtV/UFdTtVp7afiNFoFj4bXKnrEpFtCtdssg37XGsRMgaiSWuDotjH1oDes7pbw9nvPgxY4khD3Xs9mEowqVkHi0mlsYf47AQDDQJEDAAAAALBlfPTRR3r++ee/dLxQKEjSFwogQ33//El9/Gcfy67YWl1Z1eSdSRmGIbtsy5hobTbuVB3VH9RlV205tS8WOwzTUOF4od/XDy1I0juIXgnVQSR6k5Ro7ybJY9ssTDyHfc/HzTjHYFD/nQCAqChyAAAAAAAwYPmFvJyaI2vFUu1uTanJlJpuU5npjMyUKa/pybVc2VVb9ft12ZW1QkfNlWu1Xrknou3HsdWNc/I4iWNP4pjGHTEFgMGgyAEAAAAAwBAcfu6wbl652SpsTLQ2Hs9MZ2SmTcmTGk5DTs2RXWkVOtqzOqz7lo6cOTLq4X9J2E3MkzCDo99fpCclGR12nFGXawrz/Yd9z3vp92xGiWnYZyYJz1SSl/ACgCjYeBwAAAAAgCFYfGtRTs1RuVRWZbmicrGs8idllYutf1dvV1W7U9PqZ6tavbeq+v26rPuW1JRO/ejUQMcWdPPwjcdHXQiImjze/B0HlQiOun9J2HF2Oq9Xn0lIxvsRx7MZV0z7tRtmTKPExe/3AICkYSYHAAAAAABDcvbds7r4zEWVl8pyao7S2bRSmZRkSl7DU8NpyK256zM4PM/TS9deiq3/fknKXknvJNo4tqAzSzbOdujUNmgiuNuG4n7a9hJ2nP3adRIlnlGEjWnYZ3NQMe13z4PGdNhx8ds26QUwANsPMzkAAAAAABiSwvGCXnjnBVXuVrTym5XWbI5iWZVSZX02R/VOVeVSWU7F0YtXXlRuYfB7cdw6170QELTNOOmVzB5mX/36CzvOKH0mXdRncxAxTYIocfHz3ZPyPQFgI2ZyAAAAAAAwRHNPzunl+su6fOayPvzJh0pNpeQ1PUmSYRpyLVdHzhzxvUSVn6RjlMRk3EnNOK8Xx7WCXmNUsQzbNq59NaIIWziIet1BtR9WTEcRF4oYAMYRRQ4AAAAAAEbg9JundfrN01p+f1nF94qSWjM9ck8MfuYGAADAVkGRAwAAAACAEcodzSl3lMIGAABAGBQ5AAAAAGCbMgxj/b3neYHP69fez/UNw+jZN7aXIBsms6wOAACQ2HgcAAAAALaldgGiXWDYWJDwe57neT2LF/2u361PAAAAwC9mcgAAAADANtUuQHie17Pg4Pe8IO3aMzj8Xq90vaTi9bV9K44VlF/I+x4HxgezMwAAQFAUOQAAAAAAQxVkiapLi5d04/INpXekJUPSWjOn5ujwc4e1+Nbi4AYKAACAxKPIAQAAAAAYGr8FjqVrS7rw1AVl92U1f2JememMDNNQ02nKrtiyVizdvHJTr06+qrPvnlXheGEIowcAAEDSUOQAAAAAAAxVt6Wr2pauLemNZ99QfiGv2cdmlT2QVXpHWl7Tk1NxtLqyqtrdmjLTGZVLZV185qJeeOeFYX8NAAAAJABFDgAAAADYpvzuixF0/4xe7TbP4ug0s+PCUxeUX8grv5DXvm/s0865nTLTppyqo9V7q5q8M6nUZErmhClJKi+V9fqJ16V/Gmh4AAAA2AIocgAAAADANtQuPrQLEBsLDRsLD/3O2/zeT7teLi1eUnZ/VrOPzWrfN/Ypt5DTzMEZNeyGqp9WlZpKyTAMNd2mGnZDruXKqTmyHliyL9kSW3QAAABsKxQ5AAAAAGCb6lZ42Hzc73lBP+90zo3LNzR/Yl7ZA1ntnNupmYMz2j2/W9aKpYbdkFNzZJdtZaYzrVc2o3Q2rak9U7Jv2H37AwAAwNZijnoAAAAAAABIUul6SekdaWWmM0rvSMtMm2rYDVkrllzLlddcmx0yYchMmTLTpsyMqVQmpVQmJaUkYznYkloAAAAYbxQ5AAAAAGCbefzxx1UqldZfknT16tX1z0f1/udv/lwyJMM0WpuMVx1VP62qXCyrdrcmu2LLtVw13aa8pid5a8tkmWoVPgxTKgYOBwAAAMYYy1UBAAAAwDbyyiuvqFgs9lySalTvWwekptOUU2ltMp6aSsmpOTIMQ67lqv6gLqfqyLVcNZxGq+DR8FpFDwAAAGw7FDkAAAAAYJspFApfOnby5MnRv3/+pD7+s49lV2ytrqxq8s6kDMOQXbZlTLQ2G3eqjuoP6rKrtpzapmKH58krUOwAAADYTihyAAAAAAASIb+Ql1NzZK1Yqt2tKTWZUtNtKjOdkZky5TU9uZYru2qrfr8uu7JW6Ki5rT07XE/KjfpbAAAAYJgocgAAAAAAEuPwc4d188rNVmFjorXxeGY6IzNtSp7UcBpyao7sSqvQ0Z7VYd23pG+PevQAAAAYNoocAAAAAIDEWHxrUa9OvqpyqSxJci1XmWxGZsaUYbSWrHItV06ttWxV/X69VeBoSjotidWqAAAAthVz1AMAAAAAAGCjs++elVNxVF4qq1ws60HxgSrFisqlsirLFVXvVFW7U9PqvVXV7tXkeZ5euvbSqIcNAACAEaDIAQAAAABIlMLxgl545wVV7la08psVlYutYkelVGkVOW5XVb1TVblUllNx9OKVF5VbYDMOAACA7YjlqgAAAAAAiTP35Jxerr+sy2cu68OffKjUVEpes7UWlWEaci1XR84c0akfnRrxSAEAADBKFDkAAAAAAIl1+s3TOv3maS2/v6zie0VJrZkeuSeYuQEAAACKHAAAAACAMZA7mlPuKIUNAAAAfBFFDgAAAADAlmMYxvp7z/MCn9evvZ92/foGAABAdGw8DgAAAADYUtqFhnaBYXPhwc95nud1LVAYhrH+ued5HQsbvdoDAAAgPszkAAAAAABsOe0CQ6ciRJjzBql0vaTi9bX9Ro4VlF/Idzzv4Gvhrn/rXP+2t851/yxK227t+7UJotv4/PYRdnyb2w27v0H1GTWeva4X9FkJ82xFbT+IexH3MxrmXgS5D536DNsvAAwDRQ4AAAAAAGLkd6msS4uXdOPyDaV3pCVD0tqpTs3R4ecOa/GtxQGP9HMHXxtMArNbcjeu/noludufhUk+9xpf2GJTv7HGPc4wfUaNZ5D+w5wXp7hj2q1dlJjGFZcg14nynALAqFDkAAAAAAAggF6zPjYXNdpLW220dG1JF566oOy+rOZPzCsznZFhGmo6TdkVW9aKpZtXburVyVd19t2zKhwvSOqfCPWTfOyVhO2XwAya3Oz2l+N++/OrX1z8JJ/DjK/9WdBEdLfCwqDG2anPzdftd27Q/sKKK6b9hI1p2HZRYxo2LlEEfU4BYJTYkwMAAAAAgIDC7ruxdG1Jbzz7hvILeR16+pDmnpxT4VhBuSdy2v/N/dr76F7NHJrR7GOzyh7I6uIzF/XJLz4Z0LdoGXTCcvP14+wvjqJM0PHdOhc+qR4kAd7vPL9LMXU7L+hY4vzOna49isR52GczSLsoMY0jLkHuQxzPKQCMAkUOAAAAAMCW055p0W+fDb/n9WofpNBx4akL2vvIXuUX8iocK+jh7z2s/Hfz2v+t/drzyB7NHJzRrrldms5Na2d+p9IPpfX6iddDjW2U/CbYR7FM0cZ+kza+zf0ldZx+JTExHjam43wv4r4PSfyOALY3lqsCAAAAAGwp7eWk2oWLjUWIjUWJfudtft/+vNeeG5uLJRs/v7R4Sdn9Wc0+Nqt939in3EJOMwdn1LAbqn5aVWoqJcMw1HSbatgNuZYrp+bIemDp7R+8rVM/PhU+KDGIY/NjDEeY/TMG0f+o+uPZbKEYAWC7oMgBAAAAANhyus2u2Hzc73lxfHbj8g3Nn5hX9kBWO+d2aubgjHbP75a1YqlhN+TUHNllW5npTOuVzSidTWtqz5Q+eOuDgRU5om6kndSEMgnez8VRBAi6ifgon4skjMGPQT6j4xIDAIgDRQ4AAAAAAAasdL2k9I60MtMZpXekZaZNNeyGrBVLruXKa67NKpkwZKZMmWlTZsZUKpNqvaZSWn5/WbmjuUjj6JVUDbJ3wDgVEJKQ5L11rhWzTps2DyPR3e14mNiE3eB+UMb52Wwb9EbuQc4fxXMKAFGxJwcAAAAAYMt4/PHHVSqV1l9t7X/fvn1bV69eXT8+rPfF60XJkAzTkNf05FQdVT+tqlwsq3a3Jrtiy7VcNd2mvKYneWtLX5mSMdFqU3yvGDouUfTbhDiJyc8k/xV7O4ncfg1DewPrsBtZ+43nsJ+FcXw22wb5jMbxvUfxnAJAWMzkAAAAAABsCa+88oqKxWLHJaM2HhvVe3lS02nKqThavbeq1FRKTs2RYRhyLVf1B3U5VUeu5arhNFoFj4bXKnrEpFtCtdtfb/e7VhKTn0ktcHTbmHrQG1Z3KwT4vedBCxxJiHtSn822YRQ4wl57VM8pAERBkQMAAAAAsGUUCoW+xw8cOLD+/uTJk0N5XzjW6t+u2FpdWdXknUkZhiG7bMuYaG027lQd1R/UZVdtObUvFjsM01DheOfvFocgSe8kS1KivZskj22zMPHslQQfh/szaMOKQdT7sJ3vEYDxQ5EDAAAAAIAByy/k5dQcWSuWandrSk2m1HSbykxnZKZMeU1PruXKrtqq36/LrqwVOmquXKv1yj0RbT+OrW6cE+hJHHsSxzTuxj2m4z5+AFsXRQ4AAAAAAIbg8HOHdfPKzVZhY6K18XhmOiMzbUqe1HAacmqO7Eqr0NGe1WHdt3TkzJFRD/9L+m1i3mtmSNzJ0qDXG/b4wgo7zqjLNYX5/r3OHXY8B/FsRn1mhhWDJN0HABgWNh4HAAAAAGAIFt9alFNzVC6VVVmuqFwsq/xJWeVi69/V21XV7tS0+tmqVu+tqn6/Luu+JTWlUz86NdCxdUsK+zkedDPquNf0j5q4HfT4+l037KbefsfZ6bxefY5LInyUz2bQduMSUyn6cwoAo8BMDgAAAAAAhuTsu2d18ZmLKi+V5dQcpbNppTIpyZS8hqeG05Bbc9dncHiep5euvRRb//2Ssb2S3kFtnE3Q6RpxJEs3XrffX+93OhZmfH4LQnHFMuw4+7XrJEo8owgb02E/m2HaRYlplGctCjYXBzBumMkBAAAAAMCQFI4X9MI7L6hyt6KV36y0ZnMUy6qUKuuzOap3qiqXynIqjl688qJyC4Pfi+PWue6FgKBt/LRPyl+DD3N8vfoaVByj9Jl0o3o2k/5MR7WVnxkAW5fheZ43yA7Onz8vz/P0wx/+cJDdIGZb+b5t5e8GDAq/GwAAgPhdPnNZH/7kQ6WmUvKarf/X3DANuZarI2eODHyJKgAAgK2A5aoAAAAAABiB02+e1uk3T2v5/WUV3ytKas30yD0x+JkbAAAAWwVFDgAAAAAARih3NKfcUQobAAAAYVDkAAAAAAAAiRBkw2P2BwAAABIbjwMAAAAAAAAAgDHFTA4AAAAAAJAIzM4AAABBMZMDAAAAAAAAAACMJYocAAAAAAAAAABgLFHkAAAAAAAAAAAAY4kiBwAAAAAAAAAAGEsUOQAAAAAAAAAAwFiiyAEAAAAAAAAAAMYSRQ4AAAAAAAAAADCWKHIAAAAAAAAAAICxRJEDAAAAAAAAAACMJYocAAAAAAAAAABgLFHkAAAAAAAAAAAAY4kiBwAAAAAAAAAAGEsUOQAAAAAAAAAAwFiiyAEAAAAAAAAAAMYSRQ4AAAAAAAAAADCWKHIAAAAAAAAAAICxRJEDAAAAAAAAAACMJYocAAAAAAAAAABgLKVGPQAAABAfwzDW33ueF+q8bp9tPN7v8159AwAAAAAAxIWZHAAAbBHtIkO7wNCpKNHvPMMw5Hne+mtz4WLja/M1u7UDAAAAAAAYFGZyAACwhbSLD/0KDX7P66Zd1AjLa3r67P/7TMX3iqqX68p9J6f8d/NKTX35/zQ5+Fq4Pm6d69/21rnun3Vr26tNv2sEadvpGmHa97reRn6v3am9n7ZhYxFXf0H6DNo+jmel07X6tQ8a0yi/h27tB3EvRv3b8ztev/9tiuN3CwAAAKA7ihwAAGBdp9kbcbd759w7+tW//pW8pifDNGQaphrNhhr1hg6dPKTn/uI5Tc1Mhf8SARx8LXjysv1ZmCRtUHFdx8/1onyvbnHs12cv/cYa9zjD9hs1pn6vFea8OIWNadB2o/ztRXnmAAAAAIwORQ4AALBu8wyNTjM2/BzrdE7t05r+9Nt/Kq/h6Svf/ooemnlIZtpUw2nIqTqy7lta/vWy/sX+f6E//PM/1NGzRyX1T577STz2Sqb2Sl6Gbbe5/SiS0t30i6ef5PPGz/3GI2wsuhUVBjXObv1uvna/c8P0GVRcMe0nbEzDtosjnlF+e37vvZ9iC4URAAAAYPDYkwMAAAzc6r1V/dl/9WfK7svq609/XV/9O19V4cmCck/k9JXDX9HsY7Pa8/U9mn10VjMHZ/Tv/vt/p1//778e6Jj6JeSDFD6CtPdrEElSP0WIoNfwk2wOm1QPtdHzjwAAIABJREFUkgDvd16QZZWC3Ps4Yuqn783XHUXyPExMg7aLGs+wsQlz7wEAAAAkA0UOAAC2kPaSUf322fB7Xlz+fOHPNblrUnP/9ZwKxwqt//1uQfu/vV97H92rma/NaNfDuzSdm1b2QFYP7XtIf/WP/korH68MZXxJlZTEqt8E8LBnrGzuL6nj9Csp93ujsDEd93sRxVb8TgAAAECSsVwVAABbRHtfjHbhotvyUb3O67e3RrcNx3u1+w9/8h9kl20VThY0+/isDnzngGYfnZXneardranyUEWGYajZaKphN+TWXTmrjuoP6nrz77+pP/7gj6OEZSyRJB2+US4vNOriUBKLK8M0qHu/3eMKAAAADAtFDgAAtpBuG35vPt5rY/C4P/vln/5S07lpTeemtbOwU7vnd2vm0Izsiq2m25RTc2RXbGXKGWWyn7+mdk9p5eMVffrhp/rKt7/Std+wom6IPagE5qiS7RRWviiOQoCfmCZh74YkjMGPYT2jFIEAAACA8UKRAwAADEyj3lD1dlX7Htun9I60JjIT8hqeVn+7qqbTlNdszQAxJ0yZKVNm2tREZmL91XAaWvqPS5GLHL2So/0SmEE3HI7DKJOqSUjo3jrXinunDaYHnejudv0ohQC/m2QPw7DjOQiDLjB2Ox7Hvi4AAAAA4keRAwCAMVcqlb7w73w+n5jjt9+7rcx0RsZEa5krd9VV7bNaa3kr05BdseVYjhpOQ17Dk9YmgxiGIcM0ZKZM3fp/bmnhv13wEYnh6ZR8j+u6o5DkxOyoYhK1GOAnpsP+bt02Tm+PY1DPdRyG+YxuhUIQAAAAsJ1Q5AAAYIydP39ejz76qE6ePPmlz/wuXTXI457nybZtNZ2mnKqj1d+uKr2clmu5MlOmGvWG6uW6nKoj13LVcBpquk01m83WdbqvjhVIt8Rot9kC/dr6aRfUqJepSlpyu9vG1MPYsLpXMcDPfQ9S4EhC3DcWOpJolAWO9rFB/OYBAAAAxIMiBwAAY+6jjz7S888//6XjhUKh4/nDPH7gvzmgv7L/SnbNlnXfUu1uTWbKlFNzZKZMeQ1vfZNxu2LLqa0VO+yGmk5TzUZTB3//YMd+4hA2eTnIpGevZHPcyd4kJdq7SfLYOgka02He73E0TjEYp7ECAAAAWwlFDgAAMDATmQlNH5hW7U5NO2Z3KP1QWl7Tk122W/tzeJ4a9Ybsqt0qdJTt/5+9uw+Sq77vPf85px9mpJ6RRiONp6cHjYKNMLZWRpIl7Ngo4mZtk/ghvpZEwkO2SCDEm7hS7F2Su7tJqL2kqNzsrc0fbMVJZR0jJxFgUMTm4TomURJffAm5FyOTXbhyjK1gEWlm0AOMpp9On8f9o6eH0dAPp08/j96vqi7OnPN7Or9zWlT9vv37/WTn3w52mAlTW394a69vY00a5AHZfm17v7ZrUNGfAAAAAMIgyAEAADrqQ7/0IT33W88pfyEvM27Kd3zZo+UghwyVl7IqOrJztkqXy4EOJ+eolC1p07s3aWLHRK9voSu6ubRRs+U1mrnSL4PRrbSz1SWbovRpu8pqVb37jtqnrb4z3V6iqtXluvp5uS8AAABgrTN73QAAALC2ffR/+6jWbVmnxX9ZVG4+p+xcVtnZrLJzWeXmcsq9kVPhQkHFS0UV3yrKWrBUypbkWq7u+tpdHW1brYHJRgOWgzyg2erg8ep771RfNHo2YffFaFRe2Lrr1dsvQZ56wrzrnerTZvP1sj+bffar9fM7AAAAAKxVzOQAAAAd9/Pf/nn9znt/R5dfvyzXcpUcSSo2FJNhGgr8QL7jyy265WWrsiW5BVefffSz2rB1Q1vqbzSoWm/gutl89fLW2kS7W1bW3+jX+9XOVfJUy9uJvogSQInazjB5q2mlT6OK2qdRA1JR+zRKvlb7M2rfRHn2AAAAAPoDMzkAAEDHDW8c1hf+2xdkxk0t/GBB2bPl2Ry5uZxy8+XZHPkLeeXmcypcLOiTX/ykdt65s+PtOnN/7QH9KPnWunqD2d2sK8zzaeZ8u+rtZ42CO53q026+M62K+uwHYSYPAAAAsJYxkwMAAHTFus3r9MDcAzrxyyd08vdPynvTkyQFQSDDMOTart7z8ffoJ//kJ5UcSTYsr9NLx/QqbyfLa0c5zZYxiM+gmbzdDPC0Wmerbe1Gvb1qYyv5CW4AAAAAvUWQAwAAdNXH/8+P62P/4WN683tv6ty3zqm0WNLU7ill9mZkJphkCgAAAAAAwiPIAQAAus4wDW1+72Ztfu/mXjcFAAAAAAAMMIIcAAAAA6qZDZJZUgcAAAAAsBaxJgQAAAAAAAAAABhIzOQAAAAYUMzOAAAAAABc7ZjJAQAAAAAAAAAABhJBDgAAAAAAAAAAMJAIcgAAAAAAAAAAgIFEkAMAAAAAAAAAAAwkghwAAAAAAAAAAGAgEeQAAAAAAAAAAAADiSAHAAAAAAAAAAAYSAQ5AAAAAAAAAADAQCLIAQAAAAAAAAAABhJBDgAAAAAAAAAAMJAIcgAAAAAAAAAAgIFEkAMAAAAAAAAAAAwkghwAAAAAAAAAAGAgEeQAAAAAAAAAAAADiSAHAAAAAAAAAAAYSAQ5AAAAAAAAAADAQCLIAQAAAAAAAAAABhJBDgAAAAAAAAAAMJDivW4AAABAI4ZhLB8HQdB0upXnq5VRr/ywdQMAAAAAgO5jJgcAAOhrlSBDJcCwOmARNl0QBMuf1flWXluZr941AAAAAADQe8zkAAAAfa8SmGgUaAibrtl6mzF3ck6zJ2clSZm9GU3tmaqabtsj0dp05v7Gec/c31yZK8urlbdWnWHqaiVvWK3WUS1/lHvrdn2drLOdzy3MO1ar3mbTrxYlfyeeRbvf0W5891p53wAAAIBuIcgBAMCSdi6JVG/Jo0blozOiLjsVNt+xw8d06vgpJdYnJEPSUlKn4GjHbTt0+KnDTbc5qm2PtDaI2UyayrVmgyNh8obVah218tfrx6hBqkZtbXc7o9bZzucWtq+i9mkr2t2nnejPTr1rUets5t8XAAAAoBsIcgAAoCuXOjIMY3mZombSNVpOicBG79RaoqpyLczskNX5Ks4+f1ZHDhxRaktKM/tnlBxJyjAN+Y4vO2fLWrB0+sRpPTz0sO557h5l9mUkNR5cDDOIWG8wtRMDka3U1+m2NurPMIPPK683e1/NDkSvLrPT7axW5+pyw6Rvts4o2tWnjUTt06j5Wu3PKP3SrjqbzQcAAAB0E3tyAACwZHWwotV0rXDyjs48e0Z//3/8vf7+P/y9znzzjJy807H6rna19uto5OzzZ/X4Zx7X1J4pXfuxazV907QyezNK70pr4n0TGt8+rrFrx7T5+s1KTaZ09NajOvfCuQ7dRVkzA49hAypn7q+dppN5w2o2uBImXSv3VUu9/u5UOxs94yjPphPvWCVNLwbOm+3TKPla6c+o/RK1zna8pwAAAEA3EeQAAKBLKjM/Gu0V8diPPabfGvstPfbJx/TNh7+pb/7GN684dzWq9Fmjvgubrl7+WkuQVXPkwBGNXzeuqT1TyuzN6JoPX6OpD05p4v0T2nTdJo1tG9OG6Q0aSY9odGpUiXUJPbr/0Uht65SredAybACg28sora6vX9sZVj++Y1H7dNCfRTtdDfcIAACAwcByVQAAdEG95ZIqXjvxmv7oE3+kjVs3autHtyo5kpRpmvIcb3nZo3P/9ZweMh/Sz3zjZ7TtwLZu3kLPrFwarPJ3RbVlp2qlW11mtWurn8nqpaxWXj92+JhSEyltvn6zttywRek9aY1tG5Nne8qfzys+HJdhGPJdX57tybVcOQVH1qKlp29/Wge/ejByn7QDA5RrX7v2PGm1/l7V14/BFQAAAADtR5ADAIA+8M9//c/6kzv+RFs/ulXj7xnXSHpEifUJ+d7b+zoULhaUSCWUfyOvr/7rr+qnnv6pXje7a2otI1UtKNFM/kbX6l0/dfyUZvbPKDWZ0uj0qMa2jWnjzEZZC5Y825NTcGRnbSVHkuVPKqlEKqHhTcN6+amXOxbkaGYj8XYMArdSVjcG4QnmvK1dQYBuv2NR9UMbwuinDdcBAACAQUSQAwCAJZVZAWGWRAqTrhl//GN/rK0f3qr0jWlted8WjWZGFUvEZOdtFS8Vlb+QVywZk2EaUiAFbqA/+vgfSQ+2rQlowtzJOSXWJ5QcSSqxPiEzYcqzPVkLllzLVeAvzSqJGTLjpsyEKTNpKp6Mlz/Dcc2/NK/07nRL7YiyaXTY683UGbasVvK2qh8Gus/cX+6Daps2d3LQuVbZrQYBOvWORdHN/uyUftiPpHKuF+8pAAAAEBVBDgAA1P4lkVZfr7dc0lc/+1WNZka16bpN5WWPdqc19kNj8qylZY+Gyv+7vmLZo6KjxOWE7Kds6ba2dUPfmZubu+Lvqampjp1//fXX9aEPfUiS9Oyzz+rAgQM1j2dPzkqGZJiGAj+Qk3eUP5+XZ3sK/EB2zpZrufJdX4EfSMHSO2BKRqycZ/bF2ZaDHFF0YpCy2mBoN/KGKVvqjwDHar0YLG7HgHWYPu32vdUbqK+0px/fAak372gzdRLUAAAAwCAgyAEAwJJeLIkU+IG++xff1babt2lkckQjUyPaOLNRG6Y3lGcFlFzZeVvJ0aSSi+UljyqfdWPrZJ+yQ97d4HnooYe0fft23XLLLe+41uwzaOV8rWMFku/4cnKOim8WFR+Oyyk4MgxDruWqtFiSk3fkWq48xysHPLygHPRok1qDlI1+hd3KgGq1vLXqa2feZvVrgKPWxtSd3rC62V/sV9NMgKMf+n1loKMf9XOAo1fvKQAAABAFQQ4AAHpo7uScEusSSowkyssexU25RVfFt4ryLO/tZY/Mt5c9iiVi5U8yJiUkY659y2b1m1dffVV33nnnO85nMpmq6Vs5v/J4ZWCl2nFmbzmtnbNVXChq6MKQDMOQnbVlxMqbjTt5R6XFkuy8LadwZbDDMA1l9lVvUzs0GryuN0AZZeC12cHyduWtpZ8G2mvp57ZV02yftvsdW2v6OcCx0tX8jAAAADA4zF43AACAXnIcR2fOnNHZs2eXPxXz8/M6ceLE8t+dOP67x/5OhlkOYvheeWC8cKGg7LmsChcLsrNLyx45by97JEPLyx7FjJg023I3oElTe6bkFJzlDeFz8zllZ7PKzmaVm8sp/0ZehYsFFd8qqnS5JDu3FOgouHKt8ie9q/tLVV0NBnkAvV/b3q/tGlSDEuDoRlkAAABAOzCTAwBwVfvN3/xNvfvd79YnPvGJd1yLx+NKpVLLf3fieHj9sAI/kGd75VkBS8se2Xlbhrm07FG29PZMAPvKZY8CBfxkoUd23LZDp0+cVnIkKTNW3ng8OZKUmTClQPIcT07BkZ2zVbpcWp7VYV22tPOOnT1p86AsMxRVs/cQdsZLr/skajvbsVxTlD5tV1mtqnfvrfZp1Hdm0AMcAAAAQD9iWAQAcNU7ffq0Jicnlz8VW7Zs0Uc+8pHlvztxfPNP3iwF5WWPrLcs5S/kr5gRULhQkPWmJeuyVZ4NUFwR7HB8BUGgINO+PR4Q3uGnDsspOMrOZd+eyXFu6dnNL83muFBQ8VJRxTfLMzqsy5bkSwcfO9jRtrV7vfxG5YVZmijq9bBaHchd3Y5O7TlQq9yw7Y/azmrp+nFAvlmN+lPqXp+GfdcHIcDR6nsKAAAAdBMzOQAA6KH0rrQcy1FpoaTCxYJiQzH5rq9kNikzbirwA7klV07OKQc6snZ5j4elYEfgBtJk43rQGfc8d4+O3npU2bNZOQVHiVRC8WRcMqXAC+Q5ntyCuzyDIwgC3fv8vW2rv9GgajsHIsMM/DYaLI+SN4yV5Tf69X61c5U81fI2e0+1NmoOk7eeqO1slK+WVvo0qqh9GjUg1ak+bfTMm+3PdvRLlGfI5uIAAAAYFMzkAACgxz5w5we0OLuo/IX8O/Z2yM3nVDhfUOFiQdZb1nKgw8k5KmVL0o29bv3VLbMvo7ueuUu5izkt/GDhHc8u/0a+PDtnLisn5+juE3crvafze3Gcub+9g8+NyqpXXyt5uyVq29tdV5i+auZ8O+rsd/Xa36s+XQvW8jsDAACAtYeZHAAA9Njnjn5Ov7n+N5WbzUmB5FqukqmkYsmYZEi+68u13PL+Dllb1kI52GGYhvSvVd6MHD0zfdO0Hiw9qON3HNcrT76i+HC8vEm8tLyvys47doZeoirMAGI3B9/bUWenB0XbUX6zZfSqP6Lm7eb9RS2zW/fWi3oH4Zm3Oz8AAADQLQQ5AADoA/d96z595cBXtHh2UU7RWQ5yGKaxvOyRU3x7E2vDMHTft+7TF5/8Yq+bjiWHnjikQ08c0vxL85p9cVZSeaZHelfnZ24AAAAAAHC1IsgBAEAfmNgxobufvVt/8OE/UClb0rqN6xQbLgc5FEie68kreirlSjLjpu574T5tfu/mXjcbVaR3p5XeTWADAAAAAIBuIMgBAECfeNeOd+lXs7+q/3jff9Q//vE/yjAMGYahQIEUSEEQaPfdu/Wp3/9Ur5sKdFQzGx6zpA4AAAAAXN0IcgAA0Gc+/aVP69Nf+rTe/N6bVyx7NH7deI9bBgAAAAAA0F8IcgAA0KfGt49rfDuBDVx9mJ0BAAAAAAjL7HUDAAAAAAAAAAAAoiDIAQAAAAAAAAAABhJBDgAAAAAAAAAAMJAIcgAAAAAAAAAAgIFEkAMAAAAAAAAAAAwkghwAAAAAAAAAAGAgEeQAAAAAAAAAAAADiSAHAAAAAAAAAAAYSAQ5AAAAAAAAAADAQCLIAQAAAAAAAAAABhJBDgAAAAAAAAAAMJAIcgAAAAAAAAAAgIFEkAMAAAAAAAAAAAwkghwAAAAAAAAAAGAgEeQAAAAAAAAAAAADiSAHAAAAAAAAAAAYSAQ5AAAAAAAAAADAQCLIAQAAAAAAAAAABlK81w0AAAC4mhmGsXwcBEHT6Vaer1ZGmHz16gUAAAAAoJ8xkwMAAKBHKoGGSpBhdcAibLogCJY/q/OtvLY6sEFwAwAAAAAw6JjJAQAA0EOVQMPqIETUdJ00d3JOsydnJUmZvRlN7Zmqmm7bI9HKP3N/47xn7m+uzJXl1cpbq85m6qpWRrNtbabsZsqP2rbV+bpdXyfrbMczr1ZWo/zN9mmr34duPYt2v6NR2xgmbzufPQAAAPoDQQ4AAIAB1+mlp44dPqZTx08psT4hGZKWqnAKjnbctkOHnzrc9jpr2fZIawPfzaSpXIsyuBs2fyOdal+9fowapGrU1na3M2qd7XxmYfsqap+2ot192on+7NS7VqvOTn9fAQAA0BsEOQAAAAZYrSWqKtdamfVx9vmzOnLgiFJbUprZP6PkSFKGach3fNk5W9aCpdMnTuvhoYd1z3P3KLMvI6nxgGaYQcR6A5TNBDrCilJfo1/wt6u9jfqz2faFbVflWrMD0avL7HQ7q9W5utww6ZutM4p29WkjUfs0ar5W+7Od/RKmzm7/+wIAAIDOYk8OAACANazWfh2NnH3+rB7/zOOa2jOlaz92raZvmlZmb0bpXWlNvG9C49vHNXbtmDZfv1mpyZSO3npU514416G7KIuyfFSYwfFmBkKbSdeOgdIwA8PNltFKn9RSr7871c5GzzjKc+nEO1ZJ04uB82b7NEq+Vvozar9E/d624/sOAACA/kOQAwCANcIwjOVP1HRRroWtF9VV+i3McwuTrl7+ZgIdRw4c0fh145raM6XM3oyu+fA1mvrglCbeP6FN123S2LYxbZjeoJH0iEanRpVYl9Cj+x+N1LZOuZoHLcMGALq9jNLq+vq1nWH14zsWtU8H/VkAAADg6sVyVQAArAGVge/K8kS1BrTrpVudJ+y1lWnQnJXPofJ3RbVlp2qlW11mtWvVlrVafVxJc+zwMaUmUtp8/WZtuWGL0nvSGts2Js/2lD+fV3w4LsMw5Lu+PNuTa7lyCo6sRUtP3/60Dn71YPROaQMGYde+Xu+f0OvgUD8GVwAAAIBeIcgBAMAaEXYfhlrp6v3Kv52bWeffyOvct87p9edelyTN7J/R9N5ppSZTbatjkNTq29Xnw6Zrx7VTx09pZv+MUpMpjU6PamzbmDbObJS1YMmzPTkFR3bWVnIkWf6kkkqkEhreNKyXn3q5Y0GOZjYSb8cgcJhftm97pPH+Bp0akCaY87Z2BQG6/Y5F1Q9tCKOXG663c8k1AAAA9DeCHAAAYFnYX/5HCXqUsiU99snHNPutWZkxU7FETEEQ6IX/6wX5vq/pm6b108/8tBLrE9FvAC2bOzmnxPqEkiNJJdYnZCZMebYna8GSa7kK/KVZJTFDZtyUmTBlJk3Fk/HyZziu+Zfmld6dbqkdUTaNDnu9mTrD1FUJdLSzLc3oh0HZegGfTg501yq71QHrTr1jUXSzPzulk/0V5XvbSj4AAAD0H4IcAABgWa3lkhpda+S7f/pdPXXbU9pwzQbNfHRGyZGkDNOQV/Jk52xZC5Yufuei/v2Gf687/uwObf/U9vbcUB+bm5u74u+pqamOnf/e976nH/mRH5EkPfvsszpw4EDN49mTs5IhGaahwA/k5B3lz+fl2Z4CP5Cds+VarnzXV+AHUrAUADMlI1bOM/vibMtBjig6Mfhba5ZGr/Xzr857MQjfjkBAmD7t9r3V2si90o5+fT+l3r6jUfuln/sTAAAAtRHkAAAAHfWdp7+jP/+5P9c1P3yNxq8b10h6RIl1CfmeXw5wvGWpcLGgRCqh/Bt5PX3X0/rcH3+u183uqIceekjbt2/XLbfc8o5rzS5LFeZ8s8cKJN/x5eQcFd8sKj4cl1NwZBiGXMtVabEkJ+/ItVx5jlcOeHhBOejRJrUGGhvNFmhlgLLWklP1Bj7r1dsob1T9GuCotTF1pzesrhcICNv/zQQ4+qHfVwY6+lE3+yrqd6+b31kAAAB0FkEOAADWiMrsikYbgNdKV292RrMzNyo8x9Oxw8e09aNblb4xrS3v26LR6VHF4jHZeVvFS0Xlz+cVS8ZkmEZ5cN3z9cRPPCH9u6arGyivvvqq7rzzznecz2QyVdO3cn7l8crASrXjzN5yWjtnq7hQ1NCFIRmGITtry4iVNxt38o5KiyXZeVtO4cpgh2Eayuyr3qZ2CLsPRjVRBl7DLMMUdc+OKPppoL2Wfm5bNc32abvfsbWmH/og6nevE99ZAAAAdB5BDgAA1oBK0KISuKi1tFS9dPU2Iq93beX51eU++dknNXrNqDa9e5O23LBFU7unNPZDY3IsR/k3ysGNIAjku74825NruXIKjkqjJVlPWNLtrfcNmjO1Z0pOwZG1UJ5hEx+Ky3d9JUeSMuOmAj+Qa7my87ZKl0uyc0uBjoIr1yp/0ru6v1TV1aAfBo+j6te292u7BhX9CQAAgF4gyAEAwBoRdjmjejMyolyrdd53fX3/r76vmZtnlJpMaSQ9otFrRjUyNVLexLroys7Z5Q2uUwkl1i99UgkNbRqS9V1LhurPSkFn7Lhth06fOF0ObMTKG48nR5IyE6YUlGfoOAVHdq4c6KjM6rAuW9p5x86etHlQlhmKqtl7CDvjpdd9ErWd7ViuKUqftqusVtW791b7NOo70y/vFAAAAK4+Zq8bAAAA1qa5k3OKD8eVTCWVWJeQETPkFl0VLxXlFBwFXrC8wbUZM2UmTMUSMcWS5Y+RMBSca98eDwjv8FOH5RQcZeeyys3nlJ3NKnsuq+xs+e/8G3kVLhRUvFRU8c2iSpdLsi5bki8dfOxgR9vW7n0IGpUXpr5aadrV1lYHj1e3o1N7OTTqh0btj9rOaunWwoB8mPeqW30a9nvSD0Geetfb8X0HAABA/2EmBwAAA2xkZETXX3+9Xn/99eVzMzMzknTFuVrnX3vtNR04cECS9LWvfU2f+tSn2nY899Jc+ecUppb3ccifz8sreQqCQHbOllt05Tt+ecPqQJKhtwMfpqlgjiBHr9zz3D06eutRZc9m5RQcJVIJxZNxyZQCL5DneHIL7vIMjiAIdO/z97at/kaDje0cTA0zsFlvc+tGZbTS1pXlNltHo/Y1CgA0Ot/snii1RG1n2P5frZU+jSpqn0YddO9UnzZ65s32Z6fftXbmAwAAQP8iyAEAwAB74IEHdOHCharX1q1b1/D8+Pj48vHk5GRbj+NDccmXvJInO2ercKmg2FBMTt6RYRpyLVel7NLm1ZYjz/bKAQ8vUOAHCoJAQZwgR69k9mV01zN36dH9j8patDS8aVjxZFxGzFDgl/dRcS13eQbHvc/fq/Sezu/F0e7Bx0bLHjWqr3K9mYHkbqp1f51oWyt1Rc3bzfvrtnrv5tXep1G/t61+3wEAANCfCHIAADDgJiYmIp9febx37962Hmf2ZpZnbBTfKip5PilJskdtGTGjPLuj4JQ3r84ubV5dcpeDHX7gy5hmT45emr5pWg+WHtTxO47rlSdfUXw4Xp51Iy0HqnbesTP0ElVhB2bbLWygopN19LLcZstopc5e5O3m/UUts1v31ot61/r7AgAAgP5HkAMAAHTEu3a+S17Jk/WWpeLFouJDcQVeoNJiSbFETIEfyLXKyx1VAh123pZTdOQWXcmTgglmcvSDQ08c0qEnDmn+pXnNvjgrqTzTI72r8zM3AAAAAACohyAHAADomF1379KpPzml5GhSRsyQZ3tKpBKKJWKSJM/x5BQc2blyoKO0WJKds2VlLWl3jxuPd0jvTiu9m8AGAAAAAKB/EOQAAAAd8xOP/oROHT+lxdlFBUF55kYilVAsGZNhGMv7OjgFR6VsqRzoeKuk+FBc7mfc8mbkwBrSzEbSLKsDAAAAAI2ZvW4AAABY2z7/0ucVOIEW/2VR2dns25+5rHLzOeXP51W4WFDxUlH5i3mZCVOff+nzvW42AAAAAAAYAMzkAAAAHbXp3Zt073+5V1+66Uu6/PplJVNJxdfHZZqmgiBY3oDcKTg7UkmBAAAgAElEQVRKppL6+Rd/XhtmNvS62UBHMDsDAAAAANqLmRwAAKDjNr17k/7txX+rPT+/R3bRLs/kmM8q90ZOufmcXNvVvi/s0y+f/2UCHAAAAAAAIDRmcgAAgK659bdv1a2/fatycznNvjgrScrsy2gkPdLjlgEAAAAAgEFEkAMAAHTdyNSIrv/M9b1uBgAAAAAAGHAsVwUAAAAAAAAAAAYSQQ4AAAAAAAAAADCQCHIAAAAAAAAAAICBRJADAAAAAAAAAAAMJIIcAAAAAAAAAABgIBHkAAAAAAAAAAAAA4kgBwAAAAAAAAAAGEgEOQAAAAAAAAAAwEAiyAEAAAAAAAAAAAYSQQ4AAAAAAAAAADCQCHIAAAAAAAAAAICBRJADAAAAAAAAAAAMJIIcAAAAAAAAAABgIBHkAAAAAAAAAAAAA4kgBwAAAAAAAAAAGEjxXjcAAAAAa5NhGMvHQRBESteJawAAAACAtYOZHAAAAGi7SpChEmBYGXQIm84wDAVBsPxpxzUAAAAAwNrCTA4AAAB0RCVw0SjQUCtdvRkYUa/VMndyTrMnZyVJmb0ZTe2Zqppu2yNNFy1JOnN/47xn7m+uzJXlNZM3bL5a7W22nWHbEqWOavnD5F2dr9v1dbLOdj63Zt6xZvu01e9Dt55Fu9/RqG1sps5aZbTzuwsAANBPCHIAAACgb3V6Sapjh4/p1PFTSqxPSIakpWKcgqMdt+3Q4acORyo3im2PtDbw3c589dJVrrU6YNpqHbXy1+vHTvRbo+cWpZ1R62znc2vHu9Ip7e7TTvRnp961enWGyU+gAwAArEUEOQAAANC3VgYvKstQhbnW6PzZ58/qyIEjSm1JaWb/jJIjSRmmId/xZedsWQuWTp84rYeHHtY9z92jzL6MpMYDmmEGEOsNpvbbIGSn29qoP8MMPq+8HrZtlWvNDkSvLrPT7axW5+pyw6Rvts4o2tWnjUTt06j5Wu3PdvZLJ98bAACAQcaeHAAAAFiT6gU4Hv/M45raM6VrP3atpm+aVmZvRuldaU28b0Lj28c1du2YNl+/WanJlI7eelTnXjjX0bZGWYYm6uBw2EBMrXTtGiAPMzDcbBlhBn7b2W+damejZxXl2XTqHYvSp+3QbJ9GyddKf0btl1a+e1HfGwAAgEFHkAMAAAAdUVlOqtHG37XS1csXpsxaS1gdOXBE49eNa2rPlDJ7M7rmw9do6oNTmnj/hDZdt0lj28a0YXqDRtIjGp0aVWJdQo/uf7Rufd0WdbByLQxyhh3I7fav1lfX16/tDKsf35WofTrozwIAAAD1sVwVAAAA2q6yiXglGFFraal66eptRF7vWrWgSeX6scPHlJpIafP1m7Xlhi1K70lrbNuYPNtT/nxe8eG4DMOQ7/rybE+u5copOLIWLT19+9M6+NWDrXdOCzq9Fwd6r117nrRaf6/q68fgyiDo9XsDAADQSwQ5AAAA0BG1ZlKsPl9v0/Ao1+rlOXX8lGb2zyg1mdLo9KjGto1p48xGWQuWPNuTU3BkZ20lR5LlTyqpRCqh4U3DevmplzsW5AgzsNyNZap6UV69OtC+IEAn37F26oc2hNHLDdfD9A3BIwAAcDUhyAEAAICrwtzJOSXWJ5QcSSqxPiEzYcqzPVkLllzLVeAvzSqJGTLjpsyEKTNpKp6Mlz/Dcc2/NK/07nRL7YiyaXTY6+3OJ1Vvb7cGTPthYPbM/eU+qLbZcycHumuV3WoQoFPvWBTd7M9O6XawL+y+HLXO98N3CgAAoN3YkwMAAABtNTc3d8WnmfMvvvji8vlnn322rcezJ2clQzJMQ4EfyMk7yp/PKzubVeFiQXbOlmu58l1fgR9IwdKSV6ZkxMp5Zl+cbbV7Ium3Zaq6Mbjfj4OxlWBH5dMNlY2oW9ngO0yfdjvA0Ggj934OePTyHQ3bL+14bwAAAAYFMzkAAADQNg899JC2b9+uW2655R3Xml1eavUeHO04ViD5ji8n56j4ZlHx4bicgiPDMORarkqLJTl5R67lynO8csDDC8pBjzapNeDYaLZAr5apqpa/Vltb1a8BjlobU3d6w+pagYBm+r+ZAEc/9Hvl/vpVN/sq6nevHe8NAADAICHIAQAA+l6tDabDpquXf+W11WlWX6tXN9726quv6s4773zH+UwmUzX9yvMrj1cGStpxnNlbLtvO2SouFDV0YUiGYcjO2jJi5c3Gnbyj0mJJdt6WU7gy2GGYhjL7qt9DOzQahKw38Ftv4DVqvno6MWDaTwPttfRz26pptk878a6sJf3QBwQrAAAA3okgBwAA6GuVQEMQLO2XYBhVgw310tUKWqy8trqcWtcxuKb2TMkpOLIWLBUuFhQfist3fSVHkjLjpgI/kGu5svO2SpdLsnNLgY6CK9cqf9K7WtuPA9X1w+BxVP3a9n5t16CiPwEAAPoXQQ4AAND3VgYrqgUqmk1XS60ASlhv/L9v6PX//LrOPHdGkrRt/zbN3DyjyRsnI5eJ9tpx2w6dPnG6HNiIlTceT44kZSZMKZA8x5NTcGTnyoGOyqwO67KlnXfs7Emboy4zNCjLEzXblrAzXnp9b1Hb2Y7lmqL0abvKalW9e2+1T6O+M/3yTtXT78t8AQAAdBIbjwMAANRRmRVSL2hy8bsX9cX3fVFfvvnL+pv/9W/02t+9ptf+5jWd+JUT+vLNX9YX3/dFvfX9t7rYatRy+KnDcgqOsnNZ5eZzys5mlT2XVXa2/Hf+jbwKFwoqXiqq+GZRpcslWZctyZcOPnawo23rpwHKRm1pV1tbHTxe3Y5ub7Qetv1R21kt3VoYkG/Un1L3+jTsu94PQZ4w12ulGYT3AgAAICpmcgAAAKj6LI5qS1mtPvft//vb+vr/9HWNbRvTzEdnlBxJyjAMebanUrYka8FSbj6nL+78oj79u5/Wrp/d1fF7QX33PHePjt56VNmzWTkFR4lUQvFkXDKlwAvkOZ7cgrs8gyMIAt37/L1tq7/RIGW/DEKGGUxtpa0ry2/06/1q5yp5quVtFABodL7ZvU1qidrORvlqaaVPo4rap1EDUp3q00bPvNn+7PS7FuV7AQAAsFYR5AAAAIjohd95Qd948Bva+sNbNX7duEbSI4qvK+/zYOdsFd8sqnCxoMT6hJJvJPX1+78uz/Z63eyrXmZfRnc9c5ce3f+orEVLw5uGFU/GZcQMBX4g3/XlWu7yDI57n79X6T2d34ujX4IbUuOlb/qhrbXa2Im2tVJX1LzdvL9uq/d+Xe192up3bxDuEQAAoN0IcgAAgL5XmUHRaJ+NsOnaIX8hr79+4K+19SNbNfmBSU28f0Kj06MyY6bsnK3CpYKSqaRiyVi5XUuD53/5S38p/c+ShjveRNQxfdO0Hiw9qON3HNcrT76i+HBcgb+0Qb1pyLVc7bxjZ+glqsIOzLZb1DJ71d52l99sGa3U2Yu83by/qGV26956Ue+gvS/tyg8AADBoCHIAAIC+VglaVAIXK5eLWrl8VKN0q49rlbPS6mDJyjTHf+q4Rq8Z1di1Y9r83s2a3DWpTdduklNwlJvPyUyYCvylpY8sV07RkVNwlHwrKeuYJf0PkbsEbXToiUM69MQhzb80r9kXZyWVZ3qkd3V+5gYAAAAAoHUEOQAAQN+rFoCodj5suqjlV7hFVz/45g+07eZtSk2klHpXSqPpUa3fsl7WgiU7byuRTSiRSiix/srP0MYhWa9ZEqtW9ZX07rTSuwlsAAAAAMCgIcgBAADQpNmTs4oPx8sbVg/HZRiG7LytwoWC3JIr3/GloDwTxIyZMuOmYolY+ZOMyYgb0myv7wJor2Y2OmY5HQAAAADtYva6AQAAANWk02ndfPPNOn369PKnovL3sWPHls89+eSTXTv+y0f/UqZhSobkOZ7snK38+bwWZxdVuFhQabEkt+jKsz35ni/5kgJJRnm/B9M0pfkInQIAAAAAAK7ATA4AANCXPv/5z2thYaHqtc2bN0uSPvCBDyyfu/HGG7t2/EM3/JC+7X9bXslb3mQ8lozJztsyDENeyVMpW5KTd+QWXbm2K8/1FHiBAj8oL4PFxuNYY5idAQAAAKAXCHIAAIC+NTY2Vvf8yus33HBD1473fmavTv67kyplS7LespQ/n5ckJbNJmXFTvuvLKTgqLZZk52w5BUeu5cotLc3uCHwpE6IDAAAAAABAXQQ5AAAAmrT5+s2KxWOy3rJUuFCexeG7vpIjScUSMQV+INdyZeftcqAja8vJLc3qsFzJlILx+puhAwAAAACAxghyAAAANMmIGdpz3x6d/NJJJUeSMkxDbsldDnJI5b063IKrUq6k0uWSStlysKOULUl7JRm9vQcAAAAAANYCghwAAAARfOK3P6GXH39Zi/+yqCAoz9xIjCTKQQ5DCtxAbsktL1uVLQc6im8VtX58vRY/vljeiBwAAAAAALTE7HUDAAAABtUvvvKLMmKGLp+5rOxsVtlzWWVns8rN5ZR7I6f8+bwKFwoqXioqfyGvRCqhX3jlF3rdbAAAAAAA1gxmcgAAAES0bvM6feGfvqAv7fuSsrNZxZIxJVIJGTFDCiTf9WXnbPmOrw1bN+i+b92n5Eiy180GAAAAAGDNYCYHAABAC4Y3DuuXXv0l3fK/36JEKqHLr19W4UJBhYsFLZ5d1NDGIf3owz+qL3znCwQ4AAAAAABoM2ZyAAAAtMGHH/iwPvzAh6VAmn1xVpKU2ZfpcasAAAAAAFjbCHIAAAC0k0FwAwAAAACAbmG5KgAAAAAAAAAAMJAIcgAAAAAAAAAAgIFEkAMAAAAAAAAAAAwkghwAAAAAAAAAAGAgEeQAAAAAAAAAAAADiSAHAAAAAAAAAAAYSAQ5AAAAAAAAAADAQCLIAQAAAAAAAAAABhJBDgAAAAAAAAAAMJDivW4AAAAAAHSDYRjLx0EQNJ1u5flqZYQp3zCMunUDAAAAaA4zOQAAAACseZUARCXAsDpgETZdEATLn9X5Vl6rVn6tOgEAAABEx0wOAAAAAFeFSmCiVhCi2XTNqARBwpY3d3JOsydnJUmZvRlN7Zmqmm7bI9Hac+b+xnnP3N9cmSvLq5W3Vp3N1tVMnVHLWyls2dXyh8m7Ol+36+tkne185s0872b7tNXvQ7eeRbvf0ahtbKXOZvICABAGQQ4AAAAACCnskler84RNe+zwMZ06fkqJ9QnJkLSUzSk42nHbDh1+6nCzTY5s2yOtDWI2k6ZyrZmBz6gBnijlhWlfrfz1+jHqPTRqa7vbGbXOdj7zsH3V7veilTrb/Sxa6c9OvWv16qyXv5l/XwAAaIQgBwAAAACEUGuJqsq1WktUhQlwnH3+rI4cOKLUlpRm9s8oOZKUYRryHV92zpa1YOn0idN6eOhh3fPcPcrsy0hqPKAZZhCx3mBqJwYiu11fMxr1Z5jB55XXw95X5VqzA9Gry+x0O6vVubrcMOmbrTOKdvVpI1H7NGq+Vvuznf3SjfcNAIAw2JMDAAAAANqg3n4dlU/l75XOPn9Wj3/mcU3tmdK1H7tW0zdNK7M3o/SutCbeN6Hx7eMau3ZMm6/frNRkSkdvPapzL5zr6L1EmVERZnC8VpqoA8vtHCANMzDcbBmt9Ekt9e69U+1s1N9Rnmsn3rFKml4MnDfbp1HytdKfUfulHd/bqH0DAEBYBDkAAAAAXBVqBRmipquXf+UMj9XBj9VBkCMHjmj8unFN7ZlSZm9G13z4Gk19cEoT75/Qpus2aWzbmDZMb9BIekSjU6NKrEvo0f2PRmpbp/TDoHKvhA0AdHsZpdX19Ws7w+qX571S1D4d9GcRxtVwjwCA/sFyVQAAAADWvMpyUpXAxcpAQ7Vlp2qlW11mtWth99+QyntwpCZS2nz9Zm25YYvSe9Ia2zYmz/aUP59XfDguwzDku74825NruXIKjqxFS0/f/rQOfvVgE73Qfv20/wE6oxOzZqLU36v6+jG4AgAArkSQAwAAAMBVoVbwYfX5sOnCXquX7tTxU5rZP6PUZEqj06Ma2zamjTMbZS1Y8mxPTsGRnbWVHEmWP6mkEqmEhjcN6+WnXu5YkKOZjcTbMQgctqxeDbgTWHlbu4IA3X7HouqHNoTRy4Bjv/cNAGDtI8gBAAAAAD0wd3JOifUJJUeSSqxPyEyY8mxP1oIl13IV+EuzSmKGzLgpM2HKTJqKJ+Plz3Bc8y/NK7073VI7omwaHfZ6M3VGXd+/m/phMPfM/eX+q7ZpcycHumuV3epAd6fesSi62Z+d0sn+auV7CwBAJxHkAAAAALCmzc3NXfH31NRUx86fPXtW+/btkyQ9++yzOnDgQM3j2ZOzkiEZpqHAD+TkHeXP5+XZngI/kJ2z5VqufNdX4AdSsLQslikZsXKe2RdnWw5yRNGJwd9qg/adrjOMfv61ei/6pB2BgDB92u17q7WRe6Udjd7PXurlO9rP/QIAuHoQ5AAAAACwZj300EPavn27brnllndca3ZZqjDnfd+ver7WsQLJd3w5OUfFN4uKD8flFBwZhiHXclVaLMnJO3ItV57jlQMeXlAOerRJrQHKRrMFWhnYrJa3Vn3tqjOKfg1w1Nq0udObOdcLBIQd7G4mwNEP/b4y0NGPutlXzX5vAQDoFoIcAAAAANa0V199VXfeeec7zmcymarpWzm/8nhlYKXacWZvOa2ds1VcKGrowpAMw5CdtWXEypuNO3lHpcWS7Lwtp3BlsMMwDWX2VW9TOzQavK438Btl4DXMYHm766ynnwbaa+nntlXTbJ9283kPon7og2aDXAAAdAJBDgAAAADogak9U3IKjqwFS4WLBcWH4vJdX8mRpMy4qcAP5Fqu7Lyt0uWS7NxSoKPgyrXKn/Su7i9VdTXoh8HjqPq17f3arkFFfwIA8DaCHAAAAADQIztu26HTJ06XAxux8sbjyZGkzIQpBZLneHIKjuxcOdBRmdVhXba0846dPWlzL5YZ6madzZYXdsZLrwejo7azHcs1RenTdpXVqnr33mqfRn1n+uWdqmdQvhcAgLXB7HUDAAAAAOBqdfipw3IKjrJzWeXmc8rOZpU9l1V2tvx3/o28ChcKKl4qqvhmUaXLJVmXLcmXDj52sKNta/c+BI3K64d9D1odeF19D526p1rlhm1/1HZWS7cWBuQb9afUvT4N+z3phyBPmOvV0vTDdx0AsLYwkwMAAAAAeuie5+7R0VuPKns2K6fgKJFKKJ6MS6YUeIE8x5NbcJdncARBoHufv7dt9TcacGznYGqYwc1eDYavbFujX+9XO1fJUy1vs3uM1NpQPEzeeqK2s1G+Wlrp06ii9mnUgfdO9WmjZ95sf3b6XWvn9wIAgGYxkwMAAAAAeiizL6O7nrlLuYs5LfxgoTybYzar3FxueTZH/kJe2bmsnJyju0/crfSezu/Fceb+9g5CNiqr3fV1W73B7G7WFaafmznfjjr7Xb3296pP+0Wr39tBuEcAwOBjJgcAAAAA9Nj0TdN6sPSgjt9xXK88+Yriw3EFfiBJMkxDruVq5x07Qy9RFWYAsZuD772ss5vlNFtGK3X2Im837y9qmd26t17UO2jvS7vyAwDQCEEOAAAAAOgTh544pENPHNL8S/OafXFWUnmmR3pX52duAAAAAIOIIAcAAAAA9Jn07rTSuwlsAAAAAI0Q5AAAAACwzDCM5eMgCJpO1yh/q9eBsJrZSJrldAAAAAYXG48DAAAAkPR2gKESXFgZcAibLgiCmsGJRuUbhrGcnwAHAAAAgDCYyQEAAABgWSW4EARBzSBHM+nC5qsEOJoxd3JOsyeX9q3Ym9HUnqmm8mNtY3YGAADA1YEgBwAAQA+1ujRQvWurB57rXWtUP9ANYb8Pxw4f06njp5RYn5AMSUtJnYKjHbft0OGnDne4pQAAAAD6BUEOAACAHlm5dI9hGDV/yV4v3eo8q/+uNVBcL1AC9Eq9d1mSzj5/VkcOHFFqS0oz+2eUHEnKMA35ji87Z8tasHT6xGk9PPSw7nnuHmX2Zbp9CwAAAAC6jCAHAABAD7W6NFA7Zl80WibId3z941f+Ud9/5vt64/97Q5I0eeOkrvux67TrZ3bJjLPNGzrv7PNn9fhnHtfUniltvn6zUpMpJdYnFPiBnJyj4kJRhYsFJUeSys5ldfTWo7rrmbt63WwAAAAAHUaQAwAAYMBFWcoqrJe+/JKe+TfPSJIS6xJKjiQV+IF+8I0f6Pt/9X391b/5K33ydz6pG+++MWLr0W8qQa9Gs3vCpmtXviMHjmhqz5Sm9kxpyw1bNDo9KjNhysk7Kr5Z1NCFIcWH4jJj5aBb9mxWj+5/VPrVpqoBAAAAMGAIcgAAAAy4Wkv8VAt4hDlX8bVf+JpefvxlTe6cVGoyVV4aSIbckqvSYknFt4oqXirqa1/4mua+Pacfe+TH2nxn6LaVS6JV/q5Y/W7VS7f6OEy+erOUjh0+ptRESpuv36wtN2xRek9aY9vG5Nme8ufzig/HZRiGfNeXZ3tyLVdOwZG1aMk+Zkts0QEAAACsWQQ5AAAA8A5/evef6rt//l3N7J/R5us2a2RqRPF1cfmOr9JiSYVLBQ1dGFJiOCEzburbX/62nIIjXdPrlqNVYfdxCZuumeu1rp06fkoz+2eUmkxpdHpUY9vGtHFmo6wFS57tySk4srO2kiPJ8ieVVCKV0PCmYdmn7LrtAQAAADDYCHIAAAD0UKtLAzXaTyOKs/9wVt85/h1tvXmrJndOamLHhDZcs0GGaZQDHBcKiq+LL+/F4Xu+PMfTy4+9LP20JPZ6RhvNnZxTYn15qbTE+oTMhCnP9mQtWHItV4G/NDskZsiMmzITpsykqXgyrngyLsUlY765pbEAAAAADA52iQQAAOiRSnCi0ZI/9dKtXP5ndcBj5flmlqr6+i99XRtnNmps25jGrxvX5AcmNX3TtMa3j2s0M6r1E+u1bnydhjcNa2jjUPmzYUiJVEL6y1Z7Bb0yNzd3xUeSnn322eXrvTr+T0/8J8mQDNMobzKed5Q/n1d2NqvCxYLsnC3XcuW7vgI/kIKl74qpcuDDMKXZprsDAAAAwIAgyAEAANBDQRAsf1afD5OuURmN8q1WvFTU/MvzGpka0brN68qfTeuUWJ9QYjih+Lq4EusS5c9w4u3jpU3JNS/JitAR6KmHHnpI3/jGN97xvqwOqPXiuHxC8h1fTq68yXhuPqfsbFb5N/IqXiqqtFiSk3fkWq48xysHPLygHPQAAAAAsKaxXBUAoCm1NoUNm65R/jDld2J5HgBlsydnFYvHFF8XVywRU+AHsi5bMmKGfMeXV/LKA8dLv6xfXiJoaZkgI2YomOX7OYheffVV3XnnnVecu+WWW3p/fOcteu33XpOds1VcKGrowpAMw5CdtcvvpevLyTsqLZZk5205hVXBjiBQkOGdBAAAANYqZnIAAEJbvVROrT0E6qWr9YvySrqVvyKuVn6jfQuAQbBr1y79+I//uF5++eXlT8XKc/XOf/3rX1++9sgjj7Tt+OI/XVQsHpMCyS25Ki2WlH+jvDRQ/kJepcsl2QVbbunt5YFWftdjRkzGBb6naJ+pPVNyCo6sBUuFi4XlWRzZ2axycznl38ircLGg4lvF8vuZWwp0FNzynh1uIKV7fRcAAAAAOoWZHACApqxcwqRewCFsumaE3aBZkrySp9kXZ3XuhXOSpOkPTSvzwYxiQ7G2tAVoxWc/+1nZtl312nvf+95Q59/znvcsH999991tO577uzl5rifXcmVnbRUuFmTGTdl5W4ZpyCt5snP28kCyV/LkO758r/zxfE/a2KgHgObsuG2HTp84reRIUmasvPF4ciQpM2FKgeQ5npyCIztnq3S5tDyrw7psSf9dr1sPAAAAoJMIcgAABkIzS1Q9/unH9f1nvq/4UPyKfG7J1fWfvF63//ntnWwqEEoymWzp/Mq/x8bG2nZsfLC8uXNpsaTim8XyZuKBZGdtmXFTvueXB5OzdvmTt+UUHXmWJ98uLw2kTKO7B5pz+KnDenjoYWXnspIk13KVTCVlJk0ZRnnJKtdy5RTKy1aVLpfKAQ5f0iFJrFYFAAAArFkEOQAAfaPeElVhAhxnvnlGX/lXX9GG9AbNfHRGyZFk+ZfnTvmX59aCpdf/8+v6jfhv6Ge/+bPa+pGtnbgNYKBt2LpBo+lRWZcsFTYWFEvE5Dv+8q/mAz+QVyr/an75F/NLszrsoi2NStrQ67vAWnTPc/fo6K1HlT2blVNwlEglFE/GJVMKvECe48ktuMszOIIg0L3P36vf/4vf73XTAQAAAHQQQQ4AQF+ptUn56uDH6sDHmW+e0ZOfe1JbP7RVm67bpJH0iBLrEwq8YDnAUbhYUDKVVG4+p8c//bju/IsrN9gFUN5M/CP/y0d04ldOKDGSkEzJtZd+NZ8ob+fmO77cYnkweWWgwy240selgJ/NowMy+zK665m79Oj+R2UtWhreNKx4Ml7e7N4PlmdzVGZw3Pv8vUrvSUt/0euWAwAAAOgkghwAgKaE3Rejmf0z6uWX9I5ZHNVmdvzhj/6hpvdOa3LXpCbeN6GRzIhiiZicvKPim0Xlz+cVG4rJiJXb4/u+vvKvviL9WqTmAWva3v9xr1768ku69L1LCoJgeWmgWCImGSrvvVG6cg+EwsWCJm+c1NndZ1kaCB0zfdO0Hiw9qON3HNcrT76i+HBcgb+08b1pyLVc7bxjpw4+drDHLQUAAADQLQQ5AAChVYIWlcDF6lkXqzcbr5Vu9XHl+sprYfffkKQnDz6pkckRjW8f15Ybtii9O62xbWNyS64KFwrLm437ni/PLm+o7BQdlRZKso/Z0uGmugG4Ktz3wn363Z2/q7dee0sjhZFykGMoJsM03l4ayCrP5ii+WdT49nHd+w/36qGHHup103EVOPTEIR164pDmX5rX7IuzksozPdK70j1uGQAAAIBuI8gBAGhKreDD6vNh04W9Vi/dPyk3ks4AACAASURBVP0//6SZ/TMaSY9oNDOqjTMbtWHrBlkLljzbk523ZedsJReTSqaWPuuTWrdpnez/RpADqMqQfvGVX9Sx247pn//mn5U/n1diXaIcNAwkx3LkFl2ZQ6Zu+OwNOvTEoV63GFeh9O600rsJbAAAAABXM4IcAICBNvvirBLrE0qOJBVfF5eZMOWVPFlvWXItV4G/NKvENGTGTZmJ8ieWjCmWjMlIGArmWFsHqOW2Y7fpzDfP6B9++x907oVzys5mJUmpyZRmPjqjj/zKRzRz80yPWwmEE3bGYL10ta5VW56x1vVmZisCAAAAqM/sdQMAAP0vHo9rx44dOnfu3PKnovL3/Py8/vZv/3b5fLeO51+aXw5iBH4gJ+8ofyGv7GxWhUsF2VlbruXKd/3yuu3B0kCTKRkxQ6ZhypiNtm8IcLXY9iPbdPuf3a4H5h7Qr1u/rl+3fl0PzD6g2//sdgIcGBj1lkgMm66yNGPlszpwsfKzusxa+QAAAAC0hpkcAICGfu3Xfk3z8/NVr8ViseX/Dg0NLZ/v2rFRHljybE92rrw3QHwoLifvSIbklTyVFkty8o5cy5XneOWAhxco8AMFCgj5A00wE3xhMLhW7x3VbLqwMzBW7lPVTL6V5k7Oafbk0n4jezOa2jNVNd22R5ouWpJ05v7Gec/c31yZK8trJm/UfK3mbVTeSmHLrpY/TN7V+bpdXyfrbLVPa5XVKH+zfdrq96Fbz6Ld72jUNjbKG/bfpnZ8bwEAIMgBAAglna6+5vnK8xMTE8vHN998c1eOMx/MKFAgJ+fIestS/nxekmTnbBkxQ77jyyk4Ki2WZOdsOYWlYIddDnb4gS9lQnQAAABdcOzwMZ06fkqJ9QnJkLQUH3EKjnbctkOHn+reRlLbHmlt4LuT+VrN22x5lWtRBnXr9WMn+q3Rc4vSzqh1ttqnYcuKkq6d2t2nnejPTr1r9eoEAKBbCHIAAAZaendabtGVddlS4VJB8eG4fNdXaaSkWCIm3/PllcqzPEqLJdlZW07ekVMsBzvkSsEka6MDAJqzerZGo/ONrp99/qyOHDii1JaUZvbPKDmSlGGWg/V2zpa1YOn0idN6eOhh3fPcPcrsK0foGw1ohhl8rDeY2kygYy1o1J9hBp9XXg/bj5VrzQ5Ery6z0+2sVufqcsOkb7bOKNrVp41E7dOo+Vrtz3b2S706wwR3rqZ/WwAAncV6AwCAgbfzp3YqezarwvmCcvM55eZyys5mlZ3NKv9GXvnzeRUuFVR8syjrsqVStrx8VelySfpAr1sPABg0jQIZzeY7+/xZPf6ZxzW1Z0rXfuxaTd80rczejNK70pp434TGt49r7Noxbb5+s1KTKR299ajOvXCuSg3tE2W5qaiDw60sW9TOQdIwA8PNlhEmYNDOfutUOxv1d7Pnw9TZTP2ry+3F4HmzfRolXyv9GbVf6uUjSAEA6BcEOQAAA+/gEwfl2I6yc9nyZynAUfk7/0ZehQsFFS8VZb1llQMdC6XyMiCf63XrAQDdUtlfo9HG3/XStTvAIUlHDhzR+HXjmtozpczejK758DWa+uCUJt4/oU3XbdLYtjFtmN6gkfSIRqdGlViX0KP7H226DZ0UdbCzlUHSfhlgDRsA6PYySqvr69d2htUvz3ulqH066M+iFWvxngAAvcdyVQCANeG+/3Kf/vC//0Nlz2XlFl0lUgnFkrHyoJJf3pjcKTpy8o6sBUsypJ/7rz+n3zv+e71uOgCgCyqbiFcCFysDDisDEI3SrfxvvXJWqpfv2OFjSk2ktPn6zdpywxal96Q1tm1Mnu0pfz6v+HBchmHId315tifXcuUUHFmLlp6+/Wkd/OrB1jqmRWthLw7U1+ulhXodHOrH4MpaQL8CANqJIAcAYE2YvHFSd//t3fqDH/4DlRZLGt44rNhwTIZpSIHkO75cy1UpW1oOcLxrx7uk471uOQCgW2rNpFh9Pmy6qOWvdOr4Kc3sn1FqMqXR6VGN/f/s3X+MHOed5/dP1XT3jDhDccghze6mdmZpS6JsLnepESk4t8slb+2cEiDOwSSFmFawMihcgCAXEAlyfyQbgxKguyD5Z6HL7e0iuROBO1qyxaMO+SOID8yuzYPC4LziCogU3q4vXJu7ZA8lUvSQ/WOqq6q78kdPj0bj/lE/u6tm3i+g4WZXPfU8/a1qwfh+53mehVntmN8ha9nqFOgbjuyqrcJMofOaLig/ndfUzil9+M6HiRU5/CSWN8syVUH6RXxFgCSfsTilYQx+jHPD9bTHBgCw+VHkAABsGnt/Y69+r/F7+pe/+y/14VsfdmZyqFPkkCG1nJZ+/T/9df3tC3973EMFAEBL15eU35ZXYaag/La8zLyplt2StWzJtVx57dVZJROGzJwpM2/KLJjKFXKd11ROdz+4q+KzxUjjCLNptN/jcbeL2jaqNCRzb53r3LNemz0nmejud+2oie6knrEwRhnPpCQZr17xGMX+PQAADEORAwCw6Xzzn31T3/xn39S9//eeKtcrkqTykbL2fGXPmEcGYLPot1yR3/OGtY96HJ934MABLS0trf27VCpJ0uc+W//5n//5n+uZZ56RJF29elXHjx9P5H3lekUyJMPsLK3o1B3VP6mrZbfktT3ZNVuu5arttuW1PclbvfemZEx02lTer0QucoSxlZapSnNidhwxiaMQ4Cemo/5u/TZy746jV1EpLcb5jKY5LgCArYMiBwBg09pzcI/2HKSwASBe6/dq6O7dMKgQ0eu87v/229h60PXDbny9VZ0/f16VSqVnzPzEcf05SbzvLqno1BytPFhRbionp+HIMIzOMouPmnLqjlzLVctpdQoeLa9T9IhJvwTlsNkCW2mZqrQlcfttTJ30htWDCgF+k91BChxpiPv6QkcajTJWvfoIcu8BAEgKRQ4AAAAgoI2bVEc9z287ChzhlMvlQJ93Z3FI0okTJxJ7Xz7S6d+u2VpZXtHkvUkZhiG7asuY6Gw27tQdNR81ZddtOY3PFzsM01D5aO/vEIdhyetBid9Bidew7aK2DSpNifZ+0jy2XoLGdJT3O4vSEAO/Ra40jBUAsHlR5AAAAAAyxO9SVb+4+Qv96R/8qX72o5/pwc0HkqRdT+7S/r+5X8//l89r9ldnEx8rBistluQ0HFnLlhr3G8pN5tR22yrMFGTmTHltT67lyq7baj5syq6tFjoarlyr8yoeHv1SVVtBlhOyaR17WseVVcQTAIDPUOQAAAAAMsTP0lVX/t4V/dk//TPJk6Zmp7TzV3eq3Wqr8XFD1/+X6/rgzQ905D8/oq/9g6+Ncujo4eCLB3Xzys1OYWOis/F4YaYgM29KntRyWnIajuxap9DRndVhPbR06MyhsYw57F9rR/kr71H+hXjQ6/md8TLuZHTYccaxXFOYmMZ1ragGffeoMQ37zKTlmfIrzct9AQA2B3PcAwAAAAAQn3/+9X+uD/7pByoeLurJ/+BJffFrX9S+5/ep+GxRxeeKKv5GUTN7Z/Rv/ud/o7f+w7fGPdwt7/Q7p+U0HFWXqqrdralaqap6p6pqpfPv+sd1Ne41tPLpilYerKj5sCnroSW1pZPfO5no2LZiYjJq8nhjzJKKYb/r+h1/2HH2Om8zJOSHxVMaXUyHtUtTkcfP8fXS/AwAALKNmRwAAABAQN0ZFMP22fB7XlztLr5wUXf/n7taOLGguafmNFOaUW4qp5bdUvNhU41PG2psb2hiakJGztCt927prf/oLelooG4Qs7PvndXFFy6qersqp+EoP51XrpCTTMlreWo5LbkNd20Gh+d5euXaK7H1PyxJuVUSk+vjMOyv93t91m3Tq23QPUb6bSjup+0gYcc5rF0/UWIaVtiYhi1IJRXTYfc8aDyTfta2yn8nAADpRJEDAAAACKBbfOgWIPotHzXsvI3v/bTbWPhYf+zfXv63uv1/39b8b8/rC7/2Be09tFePP/G4ZEjWsqX6J3XlpnIyTENe21Pbaatlt/TzH/9celzS03FEB2GUj5b10g9f0pvH3pT1yNLUzinlCjkZE6v3ym3Ltdy1GRyvXHtFxcXk9+IgaRlMv2WdkohjlL7Cth3l9xu1QUtybfWYDluuzO8MlzR9JwDA5kORAwAAAAio34bfGz/3e16Q4/2O/ei7P9LO/Tu1Y36Hdn1pl/b82h7NPT2nlU9XVK1U5bU9teyWnBWns8dD3e7s8zDdlPPHDkWOMdv3/D59t/ldXT5zWR/94CPlpnLy2p17bZiGXMvVoTOHfC9R5TcxG7ew14wylri+RxzXCXqNcX3vUd2ncTxj43gGR9Vv1p6XqG0BAPCLIgcAAACQcbWlmh785QMtHFvQYzsf0+TspArTBUmSmTM1MTmh3FTul1+P5ZSfzkt/LRn1YEtjIRmn3j6lU2+f0t0P7qryfkVSZ6ZH8XDyMzcAAACALKLIAQAAAGRc5XplrZhhTBhqu21Zy5ZkaG2po7bblrzO8liGacicMGXmOi/DNORVBs8uwWgVny2q+CyFDQAAAGAYc9wDAAAAALLg6tWrun79+udekvTaa6+tnTOu92//wdsyJ8zOklTNzibjtbs1VStVNT5pqLnclFN35FquWk5LXsvrLIW0WvSYMCakB8FjAqTZwhv+XwAAAMguZnIAAAAAPvz4xz+W53l69dVXP/f5+fPnx/7+zH9xRv/iX/8LuSuumo+aatxvyJwwZddsmROmWnZLdq2zB4fT+KzY0XbbartttdotaZfvUAAAAABAalDkAAAAADKu/FxZ7VZbzUdNrTxYUX5bXl7bU6FakJkz5bU8OSuO7KrdedVtuQ23U+ywW/Lanrwyy1Vhc2HDYwAAgK2BIgcAAACQcTOlGc19aU4r91dU31GXmTPVcloqzBQ0kZ/oLGNlt2TXbdmPVl+rszqcFUfaLWl63N8CAAAAAIKjyAEAAABsAr/zP/yOLn/7svLb8zIMQ27TXStySFLbacuxHNk1W81HTVmPLNk1W+6KK31zzIMHAAAAgJAocgAAAACbwIH/+IB+9fiv6ta/viWv3VmeqjBd0ERhQjIkr9WZzeE0VgsdD5tqfNrQl/79L+kvnvoLidWqAAAAAGSQOe4BAAAAAIjHt//3b2v+t+b18K8eqlapqVqpqlqpqrZUU+3jmuof11W/V1fjfkP1+3V96Wtf0rf+t2+Ne9gAAAAAEBozOQAAAIBN5KX/4yX98e/9sd7/x++r9nFNE5MTyk12/m+/a7lqNVuaeGxCv/n3flMnXjsx3sECAAAAQEQUOQAAAIBN5mt//2s68p8d0Z/+4z/Vz/74Z/r0330qSdr19C598Wtf1PN/93k9/sTjYx4lAAAAAERHkQMAAADYhHYs7NDX/8evj3sYAAAAAJAo9uQAAAAAAAAAAACZRJEDAAAAAAAAAABkEkUOAAAAAAAAAACQSRQ5AAAAAAAAAABAJlHkAAAAAAAAAAAAmUSRAwAAAAAAAAAAZBJFDgAAAAAAAAAAkEkUOQAAAAAAAAAAQCZR5AAAAAAAAAAAAJlEkQMAAAAAAAAAAGQSRQ4AAAAAAAAAAJBJuXEPAAAAAEB6GIax9t7zvMDnDWs/6LjfvgEAAACgi5kcAAAAACR9VmToFhjWFx38nud5Xt8ChWEYa8c9z/tcu0HHAAAAAKAfZnIAAAAAWNMtUAwrNPg9L0lL15dUuV6RJJWPlFVaLPU8b+GNcNe/dW5421vngl1z/fX6tR1Hn2Gvt57fa/dq76ftxnaj7i/JPqPGtN+1hrUPGtOoz+ao7kXcz2iU35zfa0R53gAA2OoocgAAAAAYu43FkkHLVV06fUk3Lt9QflteMiStnuo0HB188aBOv3M64dF+ZuGNaInvpMXd56DrdY+FST4PimPY7zBsrHGPM2yfUWPq91phzotT3DFNIp5R4xL2mQsbGwAA0EGRAwAAAMBIDJr10V2uqt+/Jen2tdu6cPyCpndPa/7YvAozBRmmobbTll2zZS1bunnlpl6ffF1n3zur8tGypOGJRT9JxEHJ1KQSkWlNbg6Lp5/k8/rjfuPYPRY0Eb3xmkmPs1efG6/r5/ygfYYRV0yHCRvTsO2ixjNsXAb1P6iQ0atd0vceAIDNhD05AAAAkAjDMNZeYc4b1t7P9dnXIX3W77sRxO1rt/XWN95SabGk/V/fr33P71P5SFnFw0Xt+fIe7Xpql2b3z2ru6TlN753WxRcu6s5P7iT0LTqCJB6D/kV+HJLo009iOOg1/BQMwibVgyTAh53nd7mhfucF/dxPn0H633jdcSTOg8Y0TLso8YwSl7D3v99xChsAAPhHkQMAAACxG+cG1huvjWC6cfNTnPJz3qD2QQodF45f0K4nd6m0WFL5SFlPfPUJlZ4rac9X9mjnkzs1uzCrx/c9rpnijLaXtiv/WF5vHnsz1NiSkoak8rj4TQCPehmljf2ldZx+peV+rxc2plm/F35she8IAMAosFwVAAAAEpHkBtbDkuPdBHqQBPynP/1UkjT39JzvNptNN2Ybi0/S54sSw87b+L5fEatX372OXzp9SdN7pjX39Jx2P7NbxcWiZhdm1bJbqn9SV24qJ8Mw1HbbatktuZYrp+HIemTp3W+9q5PfPxktMBHFtadEmL/qx2iMY6ZOr/7H1V8aiyujNO77DwDAVkeRAwAAAJtKkBkCH/yvH+ja71/Tg//vgXKFzv81dm1Xc0/O6W/8N39Dh88eTnKoqdQvdr2KEkHaDzs26PiNyzc0f2xe03untX3fds0uzGrH/A5Zy5ZadktOw5FdtVWYKXRe0wXlp/Oa2jmlD9/5MLEih5/EcpzJT7/XGlfClcLKZ+IqAoz6GQsrDWPwY1TPKEUgAABGiyIHAAAAMm19UcNvgcNtuvonz/8TPbz9UI/tfEz7juyTWTDXNrCu36vrX/3X/0o/+YOf6O/85O/ImGDpq3FZur6k/La8CjMF5bflZeZNteyWrGVLruXKa6/OKpkwZOZMmXlTZsFUrpDrvKZyuvvBXRWfLUYaR5hNo/0e99Mm6Y2h45SGhO6tc52Y9dq0OclE97DNpcPGJolnLKxRxjMpScUrqfsPAAAGY08OAAAAZFavosbGDck3LlnVrDb1D/f/QzUfNrXwWwv64te+qH1f3afi4aK+8Gtf0O5ndusLX/mCdvzqDv3iZ7/Q78//vuy6PbLvNC4HDhzQ0tLS2qtr/WfXrl1b+/zq1asjeV+5XpEMyTANeW1PTt1R/ZO6qpWqGvcbsmu2XMtV223La3uSt3rPTcmY6LSpvF8JHZcowiZ/h22WPei640o4pzmJ2y12dF+j0N3AOsmNrNefMypRns1xG+UzGsf9BwAA/jGTAwAAAInwuy9GmP0z1rdbr1fBY+Nnf/hrf6j8dF5PfPUJzR2Y0/bydk0UJuRarqxlS437jc6MgYIpwzD08K8f6o8O/ZH0cqDhZcr58+dVqVR6zoJZ/1m73e75edLv5Ultpy2n5mjlwYpyUzk5DUeGYci1XDUfNeXUHbmWq5bT6hQ8Wl6n6BGTfonKYbMF4kxwdmcn9DPuZarSlsztt2lz0ps59ysE9HtWeglS4EhD3Ic9m+M26gJHr8+C3H8AABAMRQ4AAADEblQbWA/ayLqXP/nv/0TNR0198etf1J6De1T8jaIe/5XH5bU9rXy6otonNZk5s5NU725i3XT18NZD6ceSjocIRkaUy+Whn69/f+LEiZG8Lx/p9GnXbK0sr2jy3qQMw5BdtWVMdDYbd+qOmo+asuu2nMbnix2Gaah8tPd3i8Ow5OU4ihKj7DNNifZ+0jy2XoLGNI2FrzQhBgAAbH4UOQAAAJCIcW5g3eu8ttvW+3/0vuaenlvbvHruwJx2fnGnVh6syDCNTlGj4cqpdzayblabKswUNLljUs5PHBm/zd4co1ZaLMlpOGuzbHKTObXdtgozBZk5U17bk2u5suu2mg+bsmurhY6GK9fqvIqHo+3Hgd6ynDxO69jTOq6sIp4AAGwNFDkAAACwJfziL38ht+FqaueUJrdPKr8tL8Mw5DSczubVptHZtDpvaqIwoYnJCeUmc8pN5pR/LC/dl7xfxLf8Efw7+OJB3bxys1PYmOhsPF6YKcjMd2bdtJyWnIYju9YpdHRndVgPLR06c2gsY05imaFhywGNcmmjoNfzO+Nl3MnosOOMY7mmMDGN61pRDfruUWMa9pkZ9RJVYe5/Vn4XAACkHRuPAwAAIFZzc3OqVqtrr65bt26N9f3S9SXJkCbyE5Ikt+mq8WlD1UpVK5+uyKk7atkteW2vMwNkdbNrY8KQmTNlGqY0nv2rt7zT75yW03BUXaqqdremaqWq6p2qqpXOv+sf19W419DKpytaebCi5sOmrIeW1JZOfu9komOLex+Cftdb//m4E55RE68bv2NSezkMi+Ww8YcdZ6/z0pSQDyuOZzOumA5rN854hrn/vdqleY8TAADShpkcAAAAiM13vvMdPXz4UEtLS2ufbd++XZL0ox/9SN/5znfG9r5xvyHDXN3DYcWR9QtLtbu1tQ2snZXOElXuiquW3VLbaavdakvtzrJXpmGq3fhs422M1tn3zuriCxdVvV2V03CUn84rV8hJpuS1PLWczlJj3RkcnufplWuvxNZ/lFkUcfc1TuvHNuyv93t91m3Tq+2wAsCwz4PuidJP2HEOa9dPlJiGFTamYZ/NpGI67J4HjWeUZy3s/Q8bGwAA8BmKHAAAAIjNwsJC32PdosO43u/99b3y2l5n74blpur36jJMQ3bNXlsCya7baj5qyqmvbl5tt9RyWvJcT612S95elqsal/LRsl764Ut689ibsh5Zmto5pVwhJ2PCkNf21Hbbci13bQbHK9deUXEx+b044k5ADlr2ZjMkO/t9vyS+W5S+wrYd5fcbtajP5laI6Vb4jgAApBFFDgAAAGwJpedKatktNR821fi0odxjOXltT4Xq6gbWrc4G1s3q6p4O3Q2sLVdu05XX8mTsY+Pxcdr3/D59t/ldXT5zWR/94CPlpjr3UOosLeZarg6dOeR7iSq/idm4+Ul4jrrPUV4n6DWi9DmOtqP8fmGvOarvNo5+x/W8RL0GBQ0AAMKjyAEAAIAtoTBT0K/8e7+iT//dp5rcPikzZ6rltFSYXreBtd3ZwLpZbar5sCm7asuu27JrtjQveTlmcqTBqbdP6dTbp3T3g7uqvN/ZKKV8tKzi4eRnbgAAAABIF4ocAAAA2DK+efGb+kdP/yPVPqlJRmfz8cJ0QROFzmbk3SWPustWdQsdruVKp8Y8ePyS4rNFFZ+lsAEAAABsZRQ5AAAAsGXsmN+h3z7/23rvH7wnr+3JWXE+K3IYqxtY263OJuS11UJHtanf+fu/oyv1KxITObDJBN0gGQAAAEgbihwAAADYUo79t8eUn8rrT37vT2RXbU1un9TE5IQM05DneWo7q7M5qrYcy9Hf+p/+lo7+3aO68tqVcQ8dAAAAALABRQ4AAABsOV/9r76q+d+c16X/5JJqd2tquS1N5DtLVrXsliYKE5rePa3f/T9/V8VFlkPC5sXsDAAAAGQdRQ4AAABsSeXnyzr3l+f0V//XX+lnf/wz/fW1v5Yk/cpv/Yr2/839mv+t+TGPEAAAAAAwDEUOAAAAbF2GNP9b8xQ0AAAAACCjzHEPAAAAAAAAAAAAIAyKHAAAAAAAAAAAIJMocgAAAAAAAAAAgEyiyAEAAAAAAAAAADKJIgcAAAAAAAAAAMgkihwAAAAAAAAAACCTKHIAAAAAAAAAAIBMosgBAAAAAAAAAAAyiSIHAAAAAAAAAADIJIocAAAAAAAAAAAgkyhyAAAAAAAAAACATKLIAQAAAAAAAAAAMokiBwAAAAAAAAAAyCSKHAAAAAAAAAAAIJMocgAAAAAAAAAAgEyiyAEAAAAAAAAAADIpN+4BAAAAAFljGMbae8/zQp0X9liQ/gEAAABgs2MmBwAAABBAt8DQLS6sLzj4Pc8wDHmet/baeGzQ9Te2BQAAAICtjJkcAAAAQEDd4sLGAoXf84YVJ/q16xY4gli6vqTK9YokqXykrNJiqed5C28EuuyaW+eGt711Ltg111+vX9soffZrG3Scg0Tto1d7P203tht1f0n2Ged98/OM9es36PkbhWmfxL2I+xkNO8Yg14jyvAEAgM2LIgcAAACQIX6LJZdOX9KNyzeU35aXDEmrpzoNRwdfPKjT75xOeKSfWXgjWuI77rEMOxY1aRq1j37tB8UxbNyGjTXucYbtM8775jdWST+LQfqM+15EiWea4tI9RqEDAICtjSIHAAAAMEZBZ2esP7dX29vXbuvC8Qua3j2t+WPzKswUZJiG2k5bds2WtWzp5pWben3ydZ1976zKR8uShic0/SQRByVTk0pEhr1m0mMdFk8/yef1x/2OrXssaCJ64zWTHmevPjde18/5QfsMI66YDhM2pmHbRY1n2LgM6jtou6TvPQAAyAb25AAAAADGJMzyU4PcvnZbb33jLZUWS9r/9f3a9/w+lY+UVTxc1J4v79Gup3Zpdv+s5p6e0/TeaV184aLu/ORObP33EiTxGNdMikFunet//bj69ZMYDnoNPwWDsEn1IAnwYef5XW4o6D2II6Z++t943XEkzoPGNEy7KPEcdVzieE4BAMDmRpEDAAAACKi7ZNSg/TiGnTeowOH3+htdOH5Bu57cpdJiSeUjZT3x1SdUeq6kPV/Zo51P7tTswqwe3/e4Zooz2l7arvxjeb157M1AfSRtKyct/RYARr1c0Mb+0jpOv9L4jIWNadbvRZy2wncEAAC9sVwVAAAAEEB3M/BuAaLf8lHDzlv/v+uPD2o3aAPzS6cvaXrPtOaentPuZ3aruFjU7MKsWnZL9U/qyk3lZBiG2m5bLbsl13LlNBxZjyy9+613dfL7J+MJUEhx7SmRxgQ2OkYxU8dP/+Pqj2fzM8QGAADEOaE/6gAAIABJREFUiSIHAAAAEFC/GRgbP/d7XpDj/Y7duHxD88fmNb13Wtv3bdfswqx2zO+QtWypZbfkNBzZVVuFmULnNV1QfjqvqZ1T+vCdDxMrcvhJLMeZ/I5yrVEk4flr88/Elege9TMWVhrG4Mc4NxZPe2wAAEA6UeQAAAAAMm7p+pLy2/IqzBSU35aXmTfVsluyli25liuvvTo7ZMKQmTNl5k2ZBVO5Qq7zmsrp7gd3VXy2GGkcYTaN9nvcT5ugydle548qyZqGZO6tc50Y9Nq0OclEd79rR010J/GMhTXKeCYlqXgFjc24nlMAAJAd7MkBAAAA+HTgwAEtLS2tvbrWf/bTn/507fOrV6+O5H3lekUyJMM05LU9OXVH9U/qqlaqatxvyK7Zci1Xbbctr+1J3upSWaZkTHTaVN6vhI5LFGGTlMM2IY5r+as4pfmv1btJ5O5rFLobWEfZyNpPTEedCE/q2RyFpJ/RqLEZx3MKAADSj5kcAAAAgA/nz59XpVLpuVzUxn0zxvFentR22nJqjlYerCg3lZPTcGQYhlzLVfNRU07dkWu5ajmtTsGj5XWKHjHplxgd9lfYcSZUu3/17ffcjfqNNaq0Fjj6bUyd9IbV/ZLdQeIfpMCRhrgHeTbHYZyxGhabcT2nAAAgGyhyAAAAAD6Vy+VAn584cWIk78tHOv3bNVsryyuavDcpwzBkV20ZE53Nxp26o+ajpuy6Lafx+WKHYRoqH+39HeIwLHk9KEE56sRr0ES7H2lKtPeT5rH1EjSmaXrG0igrMUj7+AAAwHhQ5AAAAAAyrrRYktNwZC1batxvKDeZU9ttqzBTkJkz5bU9uZYru26r+bApu7Za6Gi4cq3Oq3g42n4c6C0ryeNe0jr2tI4rq7Iez6yPHwAAREeRAwAAANgEDr54UDev3OwUNiY6G48XZgoy86bkSS2nJafhyK51Ch3dWR3WQ0uHzhway5iTWGYoTcvWBP0Ofme8jDuZG3accSzXFCamcV0rqmHLMUWJadhnJi3PVJp+twAAIHvYeBwAAADYBE6/c1pOw1F1qara3Zqqlaqqd6qqVjr/rn9cV+NeQyufrmjlwYqaD5uyHlpSWzr5vZOJji3uBGa/663/fFhSN+i1g4qaPN44jqSSwMNiOWz8YcfZ67ysJOQHifJs9rtG2Jj6fdbHXeSJ8rvNwjMBAACSx0wOAAAAYJM4+95ZXXzhoqq3q3IajvLTeeUKOcmUvJanltOS23DXZnB4nqdXrr0SW//DkqpxJiKjJP39tI0y1vXX97OZ8sbPum16tQ2bBB7UPkwsw45zWLt+osQ0rLAxDftsJhXTYfc8aDxH/azF0RYAAGxuzOQAAAAANony0bJe+uFLqt2vafnny53ZHJWqaku1tdkc9Xt1VZeqcmqOXr7ysoqLye/FcetcvMnnQdca1tewccQ91jAGJbNH2ZefWAX5PI4+0y7KszmofVLtRimJ3+1meGYAAEB0hud5XpIdvPbaa/I8T6+++mqS3SBmm/m+bebvBiA4/psAYLO6fOayPvrBR8pN5eS1O/+X3zANuZarQ2cOJb5EFQAAAACMAstVAQAAAJvQqbdP6dTbp3T3g7uqvF+R1JnpUTyc/MwNAAAAABgVihwAAADAJlZ8tqjisxQ2AAAAAGxOFDkAAAAAYIAgGx6zPwAAAAAwWmw8DgAAAAAAAAAAMomZHAAAAAAwALMzAAAAgPRiJgcAAAAAAAAAAMgkihwAAAAAAAAAACCTKHIAAAAAAAAAAIBMosgBAAAAAAAAAAAyiSIHAAAAAAAAAADIJIocAAAAAAAAAAAgkyhyAAAAAAAAAACATKLIAQAAAAAAAAAAMokiBwAAAAAAAAAAyCSKHAAAAAAAAAAAIJMocgAAAAAAAAAAgEyiyAEAAAAAAAAAADKJIgcAAAAAAAAAAMgkihwAAAAAAAAAACCTKHIAAAAAAAAAAIBMosgBAAAAAAAAAAAyiSIHAAAAAAAAAADIJIocAAAAAAAAAAAgk3LjHgAAABgvwzDW3nueF/i89Z8PuwYAAAAAAECcmMkBAMAW1i1QdAsTGwsWfs/zPG/tBQAAAAAAMCrM5AAAYIvrFiY8z+tb5Ahynh9L15dUuV6RJJWPlFVaLPU8b+GNcNe/dW5421vn+h/r13ZQm0Ht/baL2jbotYP20au9n7Zhv1Nc/SXZZ9SY9rvWsPZBYxrl99CvfRL3Ypy/vX79h/39xvnbBQAAANAfRQ4AABCZ3yWvLp2+pBuXbyi/LS8ZklZPdRqODr54UKffOZ3wSD+z8EbvJOSgZHD3WL/kZdiiTNS2Ua8/7HsNat8vjsP6HGTYWOMeZ9g+o8bU77XCnBenuGOatt+en77DFnQAAAAAJI8iBwAAiGRjUcMwjF/67Pa127pw/IKmd09r/ti8CjMFGaahttOWXbNlLVu6eeWmXp98XWffO6vy0bKk4QlNP4nHQcnUQcnLsO3Wtw2T9IzS1u+1Nxr2vfr9dXrS8dh4zaTH2avPjdf1c37QPsOIK6bDhI1p2HZR4xn19xP0/gMAAAAYP/bkAAAAibp97bbe+sZbKi2WtP/r+7Xv+X0qHymreLioPV/eo11P7dLs/lnNPT2n6b3TuvjCRd35yZ1ExzQsSRqk8OG3rZ8xJbm8jZ/EcNBrJBGPQQWspMY5rGgW5nkIs8SR36LdOJZBChrTMO2ixDNKXMLe/yDXAAAAAJAcihwAAGxx3aWmhu2z4fe8jS4cv6BdT+5SabGk8pGynvjqEyo9V9Ker+zRzid3anZhVo/ve1wzxRltL21X/rG83jz2Zrgvg9j5TQCP+q/dN/aX1nH6lcbkeNiYZv1ehJXGewgAAABsBSxXBQDAFtbdRLxbuFi/zNT6ZaeGnbfxml2XTl/S9J5pzT09p93P7FZxsajZhVm17Jbqn9SVm8rJMAy13bZadkuu5cppOLIeWXr3W+/q5PdPJvbdAWn8f4E/7uLQVk/Mx3H/N1uxBgAAAMgaihwAAGxx/TYK3/i53/PWu3H5huaPzWt677S279uu2YVZ7ZjfIWvZUstuyWk4squ2CjOFzmu6oPx0XlM7p/ThOx8mVuSIuiH2ZksMk6T9TFxFAD8xTcPzlIYx+DGqZzTo/c9K/AAAAIDNjCIHAABIxNL1JeW35VWYKSi/LS8zb6plt2QtW3ItV157dXbIhCEzZ8rMmzILpnKFXOc1ldPdD+6q+Gwx0jjCbBo9qO1mTmam4bvdOteJe68NppNMdPe7dtQkdpiNzpMyyngmJal4Rbn/afjdAAAAAFsZRQ4AALaopaWlz/27VCrF9vnVq1c18xczkiEZpiGv7cmpO6p/UlfLbslre7JrtlzLVdtty2t7kre69JUpGROdNpX3K5GLHHHrlXzPsjT/Jfo4kvBxFAL8xHTU363fRu7dcaT5uR7lMxrk/mexSAQAAABsRhQ5AADYgl577TU99dRTOnHixC8dC7osVa/P1z7zpLbTllNztPJgRbmpnJyGI8Mw5Fqumo+acuqOXMtVy2l1Ch4tr1P0iEm/xGi/2QLD2vpplxVpLXD025g66Q2rBxUC/N7zIAWONMR9faEjjcZZ4Oh+1uv+p+keAgAAAFsdRQ4AALaon/70p/r2t7/9S5+Xy+We5wf5/MSJE1p6vDPDw67ZWlle0eS9SRmGIbtqy5jobDbu1B01HzVl1205jc8XOwzTUPlo7z7jEDR5HbVd2mQhSZvmsfUSNKZ+ZglkLQZxykIMuIcAAADA+FHkAAAAiSgtluQ0HFnLlhr3G8pN5tR22yrMFGTmTHltT67lyq7baj5syq6tFjoarlyr8yoeTtdSVZtFlpOvaR17WseVVcQTAAAAgF8UOQAAQGIOvnhQN6/c7BQ2JjobjxdmCjLzpuRJLaclp+HIrnUKHd1ZHdZDS4fOHBr38DeloMnjYTNX0pKMDjvOOJZrChPTuK4V1aDvHjWmYZ+ZUS9RFeb+p+keAgAAAFudOe4BAACAzev0O6flNBxVl6qq3a2pWqmqeqeqaqXz7/rHdTXuNbTy6YpWHqyo+bAp66EltaWT3zuZ6Nj6JTaHJTzTvH/BMFGTrxu/e1KxGHZvho0/7Dh7nZemhHxYfp71UcXU7+9rHPEMc/8BAAAAjB8zOQAAQKLOvndWF1+4qOrtqpyGo/x0XrlCTjIlr+Wp5bTkNty1GRye5+mVa6/E1v+wpOqgvzSPq12/DbTjauvH+usM++v9Xp912/RqO6wAMOzzsPeg1zjCjHNYu36ixDSssDENW5BKKqbD7nnQeEZ51sLefwAAAADpwEwOAACQqPLRsl764Uuq3a9p+efLndkclapqS7W12Rz1e3VVl6pyao5evvKyiovJ78Vx61z/hGeYdpvdoGT2KPvyc3+CfB5Hn2k3aPzjimnabOb7DwAAAGx2zOQAAACJ2/f8Pn23+V1dPnNZH/3gI+WmcvLaniTJMA25lqtDZw75XqLKT9IxSmIybNtx9DnK6we9xrjiMar7N8oCT9Q+o451FP2O+/cT1/2kKAIAAACMFkUOAAAwMqfePqVTb5/S3Q/uqvJ+RVJnpkfxcPIzNwAAAAAAwOZDkQMAAIxc8dmiis9S2AAAAAAAANFQ5AAAYJVhGGvvPc8LdV6YY+s/D3IMW1uQDZJZPgcAAAAAsFmx8TgAAPqsmNAtImwsLvg5b9gxz/PWXr2KF93XRoOOAQAAAAAAbGXM5AAAYFW3iNCrCOH3PL/XSNLS9SVVrq/ud3GkrNJiaSzjQLKYnQEAAAAAAEUOAABGYmPRI45lrja6dPqSbly+ofy2vGRIWj3VaTg6+OJBnX7ndIRvAAAAAAAAkD4UOQAAGIHuclW9/t2rqOHnWNfta7d14fgFTe+e1vyxeRVmCjJMQ22nLbtmy1q2dPPKTb0++brOvndW5aPlJL4iAAAAAADAyFHkAAAgw25fu623vvGWSoslzT09p+m908pvy8tre3JqjlaWV9S431BhpqDqUlUXX7iol3740riHDQAAAAAAEAuKHAAArOrOkhi2l8ag8/xeIy4Xjl9QabGk0mJJu5/Zre37tsvMm3LqjlYerGjy3qRykzmZE6YkqXq7qjePvSn9dyMZHgAAAAAAQKIocgAAoM/2zOgWJwYtLdXvPD/H1v97/fU3jsXPsUunL2l6z7Tmnp7T7md2q7hY1OzCrFp2S/VP6spN5WQYhtpuWy27Jddy5TQcWY8s2ZdsiS06AAAAAABAxlHkAABgVb9NvTd+Pmjz7zDHwl7vxuUbmj82r+m909q+b7tmF2a1Y36HrGVLLbslp+HIrtoqzBQ6r+mC8tN5Te2ckn3D7ntdAAAAAACArDDHPQAAABDc0vUl5bflVZgpKL8tLzNvqmW3ZC1bci1XXnt1VsmEITNnysybMgumcoWccoWclJOMu6NZUgsAAAAAACApFDkAAFvegQMHtLS0pKWlJX388ce6evXq2rG0vq9cr0iGZJhGZ5PxuqP6J3VVK1U17jdk12y5lqu225bX9iRvdekrU53Ch2FKFT/RAQAAAAAASC+WqwIAbGnnz59XpVL5pf0zsvBentR22nJqnU3Gc1M5OQ1HhmHItVw1HzXl1B25lquW0+oUPFpep+gBAAAAAACwCVDkAABseeVy+XP/3rt379r7EydOpPJ9+UhnzHbN1sryiibvTcowDNlVW8ZEZ7Nxp+6o+agpu27LaWwodnievDLFDgAAAAAAkG0UOQAAyKDSYklOw5G1bKlxv6HcZE5tt63CTEFmzpTX9uRaruy6rebDpuzaaqGj4Xb27HA9qTjubwEAAAAAABANRQ4AADLq4IsHdfPKzU5hY6Kz8XhhpiAzb0qe1HJachqO7Fqn0NGd1WE9tKRfG/foAQAAAAAAoqPIAQBARp1+57Ren3xd1aWqJMm1XBWmCzILpgyjs2SVa7lyGp1lq5oPm50CR1vSKUmsVgUAAAAAADLOHPcAAABAeGffOyun5qh6u6pqpapHlUeqVWqqLlVVu1tT/V5djXsNrTxYUeNBQ57n6ZVrr4x72AAAAAAAALGgyAEAQIaVj5b10g9fUu1+Tcs/X1a10il21JZqnSLHx3XV79VVXarKqTl6+crLKi6yGQcAAAAAANgcWK4KAICM2/f8Pn23+V1dPnNZH/3gI+WmcvLanbWoDNOQa7k6dOaQTn7v5JhHCgAAAAAAEC+KHAAAbBKn3j6lU2+f0t0P7qryfkVSZ6ZH8TAzNwAAAAAAwOZEkQMAgE2m+GxRxWcpbAAAAAAAgM2PIgcAAFucYRhr7z3PC3Vev2PrPw/aFgAAAAAAYBiKHAAAbGHdAoPneTIMQ4Zh9Cw0DDpvY5v1/x5U0OjVFgAAAAAAIAiKHAAAbHHrCxK9Zl4MO89vkWJQMcSvpetLqlxf3W/kSFmlxVLP8xbeCHTZNbfODW9769zw6/S6hp92UduGuXaQ64cd28Z2o+4vqT6jxnPQ9YZdI2hMoz7Xo7oXcT+jST5rcfy3AgAAAEB0FDkAAEBkYZec8tvu0ulLunH5hvLb8pIhafVUp+Ho4IsHdfqd04HHHNbCG8GT3uuPhW0bNWEaZWyD2oeNxyBRYhFmnGH6jBrPIP2HOS9Occe0X7ukfj+DxH0fAQAAAIweRQ4AABDZsBka/WZtDGt3+9ptXTh+QdO7pzV/bF6FmYIM01Dbacuu2bKWLd28clOvT76us++dVfloWdLwRKifpOWgJOywpHfQtn77jWpYXPwkn9cfD/qdgn6PfjFOapy9+tx43WHnBu0vrLhiOkzYmIZtFzWmQeKS5G8WAAAAwOiY4x4AAABAL7ev3dZb33hLpcWS9n99v/Y9v0/lI2UVDxe158t7tOupXZrdP6u5p+c0vXdaF1+4qDs/uZPomMIWR4a1HVZ8iSOJGkeSduN5fgoGYZPqQRLgw87zu+xQkPjHmfQOWnwbR1I9aEzDtIsS0yhxSfJ3BwAAACB5FDkAANjiuktGDdqPY9B5w9qFdeH4Be16cpdKiyWVj5T1xFefUOm5kvZ8ZY92PrlTswuzenzf45opzmh7abvyj+X15rE3ExnLVua3ADDqZZQ29pfWcfqVxoR62Jhm/V4AAAAAyBaWqwIAYAvrbiLeLVT0Wz5q0HnDNiIftFRVv3aXTl/S9J5pzT09p93P7FZxsajZhVm17Jbqn9SVm8rJMAy13bZadkuu5cppOLIeWXr3W+/q5PdPRgnLSLHu/3iNO/7jLg7x3EVHTAEAAIDxYiYHAABbnOd5a6+Nn/s5z8+xoH3fuHxDO5/cqem909q+b7tmF2a1Y36Htu3epqnZKU0+PqnCTOGz13RB+em8pnZO6cN3Pgzy9QMZlJAe9tfpfpPpC298/jUK/EX9Z+KIf9BNxMeZFB/lcxZFEmOM6zfbq10WYgoAAABsFszkAAAAqbJ0fUn5bXkVZgrKb8vLzJtq2S1Zy5Zcy5XXXp1VMmHIzJky86bMgqlcIdd5TeV094O7Kj5bjDQOPwWNXp8PSnD62Zej3+ejSISn4S/Q18dw0ObucUsi/lH2cEnCKOOZlLjjFeU32+t4FmMKAAAAZB0zOQAA2KIOHDigpaWltVfX+s+Gff5nf/Zna8evXr0ay/sfv/1jyZAM05DX9uTUHdU/qataqapxvyG7Zsu1XLXdtry2J3mr+4KYkjHRaVN5vxIxOuPT3UB5VBtMp2E2QT/jmNUSNf5BZuyM0rDN09OcnE/rM5rlmAIAAACbCTM5AADYgs6fP69KpRJoealh58b1vvOB1HbacmqOVh6sKDeVk9NwZBiGXMtV81FTTt2Ra7lqOa1OwaPldYoeMemXUO03y6B7rF/bQe0G9TloZkNUaU8e99vrIKnkcdT4By1wpCHu3e+XVknHKupvtpe0xxQAAADYbChyAACwRZXL5cifP/fcc2vvT5w4Ec/7b5/Qz/7wZ7JrtlaWVzR5b1KGYciu2jImOpuNO3VHzUdN2XVbTuPzxQ7DNFQ+2vs7xKFf0ntYMjbJYkUYaUq095PmsW0UJp6DEuFZuD9JG2eBo/t5mn6zAAAAAHqjyAEAAFKltFiS03BkLVtq3G8oN5lT222rMFOQmTPltT25liu7bqv5sCm7tlroaLhyrc6reDjafhybXZYT6GkcexrHlHXEFAAAAIBf7MkBAABS5+CLBzt7cNxrqHa3pmql2nktVVW7W1P9Xl2N+w2t/GJFzYfNtVkd1kNLh84cGvfwAxl1Ejdo8njYElFpSUaHHWfUcYf5/hv3/ei1B8io92TpN85B5wyLadhnJi3PVFgsVQUAAACMFkUOAACQOqffOS2n4awVNaqVqqp3OoWO2t2a6h/X1bjX0MqnK1p50Cl0WA8tqS2d/N7JRMc2LIE5LLEbtG2cCd+o19o4vqSSuVELKmHHGTT+WUnG+3kmRxVTv7+fUcY0zG82jpgCAAAAiAfLVQEAgFQ6+95ZXXzhoqq3q3IajvLTeeUKOcmUvJanltOS23DXZnB4nqdXrr0SW//DkrG9/nq928bPX8eHaRvF+uvGPb5hf5E/7PNBBYQgwo4zTPyjxDOKsDEN+1wlFdNh9zxoTMPEJcpvdlgbAAAAAKPDTA4AAJBK5aNlvfTDl1S7X9Pyz5fXlqyqLdXWZnPU79VVXarKqTl6+crLKi4mvxfHoGWEwh7rHg/TblQGjW+UfQ3rL+w40x7/KIY9d+OIaVpE+T2HuSYAAACA+Bme53lJdvDaa6/J8zy9+uqrSXaDmG3m+7aZvxsAbFaXz1zWRz/4SLmpnLx25/+6GKYh13J16MyhxJeoAgAAAAAA6cRyVQAAIPVOvX1Kp94+pbsf3FXl/YqkzkyP4uHkZ24AAAAAAID0osgBAAAyo/hsUcVnKWwAAAAAAIAOihwAAAAZFGTTY/YHAAAAAABsVmw8DgAAAAAAAAAAMomZHAAAABnE7AwAAAAAAJjJAQAAAAAAAAAAMooiBwAAAAAAAAAAyCSKHAAAAAAAAAAAIJMocgAAAAAAAAAAgEyiyAEAAAAAAAAAADKJIgcAAAAAAAAAAMgkihwAAAAAAAAAACCTKHIAAAAAAAAAAIBMosgBAAAAAAAAAAAyiSIHAAAAAAAAAADIJIocAAAAAAAAAAAgkyhyAAAAAAAAAACATKLIAQAAAAAAAAAAMokiBwAAAAAAAAAAyCSKHAAAAAAAAAAAIJMocgAAAAAAAAAAgEyiyAEAAAAAAAAAADKJIgcAAAAAAAAAAMik3LgHAIyTYRhr7z3Pi3SeYRg9j/Vr67dvAAAAAAAAAEBvzOTAltUtMnQLDOuLDkHPG9TW87y1V7/P+7UHAAAAAAAAAPTHTA5sad3Cw7BCw6DzugWLfp8Pul4QS9eXVLlekSSVj5RVWiz1PG/hjcCXliTdOje87a1zwa65/nr92vbrM0hfva4RdKxBrh3k+mHHtrHdqPtLss847nmvaw1rHzSmUX8Po7oXcT+jYcfop23c/40BAAAAAACgyAFEMKiQ0T3etfE8v8tVXTp9STcu31B+W14yJK2e6jQcHXzxoE6/czrc4ENYeCNa4jvIOd1jYZK73WNRE6ZJjW/Q2MIWqaLEIsw4w/YZNaZ+rxXmvDjFHdMk4pnUszaoTwAAAAAAgLhR5ABCGlbgkH55D471/x50TJJuX7utC8cvaHr3tOaPzaswU5BhGmo7bdk1W9aypZtXbur1ydd19r2zKh8tSxqe0PSTfByUTI2jeDCsr6D9bTwe51jDjq/fDAO/Y+seC5qIDhqLqOPs1efG6/o5P2ifYcQV02HCxjRsu6jxDBOXuPoEAAAAAACIij05gAgMw1h7df8dh9vXbuutb7yl0mJJ+7++X/ue36fykbKKh4va8+U92vXULs3un9Xc03Oa3jutiy9c1J2f3Iml737CLB8VJjnut79BfcSVQI0jSbvxPD8xCZtUjxKLoOMcdo+Dfu6nzyD9b7zuOJLqQWMapl2UeIaNC8ULAAAAAACQJhQ5sKX5LU70Om/9xuHr9+wI0m8/F45f0K4nd6m0WFL5SFlPfPUJlZ4rac9X9mjnkzs1uzCrx/c9rpnijLaXtiv/WF5vHnvTV9+jkpZk5ziXKhqW6B/12Db2l9Zx+pWWZ2y9sDHN+r0AAAAAAAAYF5arwpbV3Sy8W3Dot3zUoPP8XH/9v/0cu3T6kqb3TGvu6Tntfma3iotFzS7MqmW3VP+krtxUToZhqO221bJbci1XTsOR9cjSu996Vye/fzJENOJDEnbzG/e+C+MuDqWxuJI1xBQAAAAAAMSFIge2tH4Fi42f+yls9DpnULt+x25cvqH5Y/Oa3jut7fu2a3ZhVjvmd8hattSyW3IajuyqrcJMofOaLig/ndfUzil9+M6HiRU5gmwkHkfCMu3FkrSPb5TiSliP+hkLKw1j8CNNG677bZf2mAIAAAAAgPShyAGkyNL1JeW35VWYKSi/LS8zb6plt2QtW3ItV157dVbJhCEzZ8rMmzILpnKFXOc1ldPdD+6q+Gwx0jjCbBrt93hQg5bvWXij9wbHo0zupiEpO65Y9Lt21IT1qJ+xIH1lsbiVhv1IBh3LYkwBAAAAAEB6UOTAlnTgwAEtLS2t/btUKknS5z4L+vmdO3d05MgRSdLVq1d1/PjxwO8r1yuSIRmmIa/tyak7qn9SV8tuyWt7smu2XMtV223La3uSt7q/hykZE502lfcrkYscYcSZqAyaJB/X8kVpKHBsNI6EcRxJaz8xHfV367eRe3ccvYpKaTGOZ9RPn1mOKQAAAAAASCeKHNhyzp8/r0qlEmh5qSifB30vT2o7bTk1RysPVpSbyslpODIMQ67lqvmoKafuyLVctZxWp+DR8jpFj5j0SzIOmy0Q5zJVfq7VbzPmJDdpTmuBYxyxWH/9jZ/1e1Z6CVLgSEPc1yfl0yitBY5B0h5TAAAAAACQXhQ5sCXl5v4jAAAgAElEQVSVy+XYP1///sSJE6Hel490rmHXbK0sr2jy3qQMw5BdtWVMdDYbd+qOmo+asuu2nMbnix2Gaah8tPdY4zAseT0oSZlkIntUydw0Jdr7SfPYeolz1k4W7k/SsljgAAAAAAAAiIIiB5AipcWSnIYja9lS435Ducmc2m5bhZmCzJwpr+3JtVzZdVvNh03ZtdVCR8OVa3VexcOjX6oqDnEnStN+vVFK69jTOq6sosABAAAAAAC2IoocQMocfPGgbl652SlsTHQ2Hi/MFGTmTcmTWk5LTsORXesUOrqzOqyHlg6dOTSWMUednZH2RGnQ8fmd8TLu7xt2nHEsLRQmpnFdK6pB3z1qTMM+M1kvcLBUFQAAAAAACMsc9wAAfN7pd07LaTiqLlVVu1tTtVJV9U5V1Urn3/WP62rca2jl0xWtPFhR82FT1kNLaksnv3cy0bGlbY+LfuMZ1x4hg9r3+3dcosYi7Dh7nZfGhHxQw+IpjS6mw9plpcARR0wBAAAAAAA2YiYHkEJn3zuriy9cVPV2VU7DUX46r1whJ5mS1/LUclpyG+7aDA7P8/TKtVdi639YUjXuJaCG9ennr/iTEGV862c79Go7rAAw7POge6L0E3acw9r1E8c9DypsTMM+W0nFdNg9DxrPOOISV58AAAAAAABhMZMDSKHy0bJe+uFLqt2vafnny53ZHJWqaku1tdkc9Xt1VZeqcmqOXr7ysoqLye/Fcetcuv7SelCyOA3jHDS+UfY1rL+w40x7/KMYNP5xxTTrosYUAAAAAACgF8PzPC/JDl577TV5nqdXX301yW4QM+5belw+c1kf/eAj5aZy8tqdn6thGnItV4fOHEp8iSoAAAAAAAAASCuWqwJS7tTbp3Tq7VO6+8FdVd6vSOrM9CgeTn7mBgAAAAAAAACkGUUOICOKzxZVfJbCBgAAAAAAAAB0UeQAsGUE2fSY/QEAAAAAAACA9GPjcQAAAAAAAAAAkEnM5ACwZTA7AwAAAAAAANhcmMkBAAAAAAAAAAAyiSIHAAAAAAAAAADIJIocAAAAAAAAAAAgkyhyAAAAAAAAAACATKLIAQAAAAAAAAAAMokiBwAAAAAAAAAAyCSKHAAAAAAAAAAAIJMocgAAAAAAAAAAgEyiyAEAAAAAAAAAADKJIgcAAAAAAAAAAMgkihwAAAAAAAAAACCTKHIAAAAAAAAAAIBMosgBAAAAAAAAAAAyiSIHAAAAAAAAAADIJIocAAAAAAAAAAAgkyhyAAAAAAAAAACATKLIAQAAAAAAAAAAMokiBwAAAAAAAAAAyKTcuAcAANgaDMNYe+95Xujz+h0f1M5v3wAAAAAAAMgWZnIAABLXLTJ0Cwzriw5BzjMMQ57nrb38tNvYpl/fAAAAAAAAyB5mcgAARqJbgBhWaOh3XrdYEbRdmJkbS9eXVLlekSSVj5RVWiz1PG/hjcCXliTdOje87a1zwa65/npB2vpp5/d7Bh2z3378XrdXez9tN7YbdX9J9hk1pv2uNax90JhG/T2M6l7E/YyG/c1EuRdR+gUAAACANKLIAQDIjLDFC7/tLp2+pBuXbyi/LS8ZklZPdRqODr54UKffOR14zGEtvBEt8Z1kuyQMGkv3WJjk86A4JhG3YfctzDjD9hk1pn6vFea8OMUd0yTiGWdcglwrbGwAAAAAIEsocgAAMmPjElV+Cx3D2t2+dlsXjl/Q9O5pzR+bV2GmIMM01Hbasmu2rGVLN6/c1OuTr+vse2dVPlqWNDyh6SeJOCiZmpZEpJ9EcRzjHBZPP8nn9cf9xrF7LGgieuM1kx5nrz43XtfP+UH7DCOumA4TNqZh20WNZ9i4hBHH8wYAAAAAWcCeHACALe32tdt66xtvqbRY0v6v79e+5/epfKSs4uGi9nx5j3Y9tUuz+2c19/ScpvdO6+ILF3XnJ3cSHVOY5abCJofTkuT0kxgOeg0/BYM445bUOIfdq6Cf++kzSP8brzuOZypoTMO0ixLPuOIS5ncbNjYAAAAAkBUUOQAAI9FdMmrYxt9+z/Pbbth1Lhy/oF1P7lJpsaTykbKe+OoTKj1X0p6v7NHOJ3dqdmFWj+97XDPFGW0vbVf+sbzePPZmoLElLWzSMqk9NEbJbwFg1GPd2F9ax+lXGhPjYWO6Fe5F1r8jAAAAAATBclUAgMR1NwPvFhz6LR816LxBG4qHbXfp9CVN75nW3NNz2v3MbhUXi5pdmFXLbqn+SV25qZwMw1Dbbatlt+RarpyGI+uRpXe/9a5Ofv9kLPEJKy17caQxAb5ZjHvGzbiLQzxbn6EgAQAAAAC9UeQAAIxEv/0z/n/27j9GjvO+8/ynqn/MiD1DDjmk2d2UZ0RbpGTRxJJjkmc7ZsgkutUeLr6DyRFiScA6RyEHHJCFkEP2j8ueIRHR3q9/fMJtfmySE+ELLcbk0oCBW8SBzokZKPStLR6xocCNlTASLap7+FMz07+qq6q77o+eHg1H091V1b9n3i+goJ6qeup56lvVFPB8+3me1fubrbPR6WPXL17X1NEpJXYmNL5rXBPTE9oytUXWvKWKXZFTdGTnbMXH4rUtEVcsEdPo1lFdO3+ta0kOP52Z62WaKj/o3P1Yp5IA3XzHOmkQ2uBHt9/RYYkDAAAAAPQDSQ4AwIaUvZJVbFNM8bG4YptiMmOmKnZF1rwl13LlVZdGh0QMmVFTZsyUGTcVjUdr22hUc1fnlDyYbKsdYRaN9nu80+VW6ken6yB08N58qXbvay3a3M2O7kbXbvc5dOsdC6OX8eyWbsZrEN5/AAAAABhEJDkAAF2VzWYf+juVSnVtfyKR0NWrV3Xs2DFJ0qVLlxp+HvvZmGRIhmnIq3pyCo4Kdwqq2BV5VU923pZruaq6VXlVT/KW1vcwJSNSK5N5O9N2kiOMQZmmqhcG+Rfs/YhnJxIBfmLa63trtJB7vR1rJZUGRbff0WH83gIAAABAL5HkAAB0zenTp7Vnzx4dP378E8f8Tl8VdP/q9TiafvakqlOVk3dUelBSdDQqp+jIMAy5lqvyYllOwZFruao4lVrCo+LVkh4d0qhjtNVogY00TdWgtbnRos3dXsy5WSLAbxIgSIJjEOK+MtExiHqV4BiEZwEAAAAAg4okBwCgq9599109//zzn9ifTqfXPL/d/SsTKs0+ZzfXRoDYeVul+ZJG7o7IMAzZOVtGpLbYuFNwVF4syy7YcooPJzsM01D68Npt6oRWndfNOn6bdYyGLRfmvHYMQ+fuILdtLUFj2ol3ZT3rZQx4FgAAAADQGEkOAMCGlJpJySk6suYtFe8VFR2JqupWFR+Ly4ya8qqeXMuVXbBVXijLzi8lOoquXKu2JQ/0fqqqjWCYO20Hte2D2q5hRTwBAAAAYHCQ5AAAbFj7nt2nG2/eqCU2IrWFx+NjcZkxU/KkilORU3Rk52uJjvqoDmvB0v7n9velzWGnGerU9ETdnjooaOex3xEv/e6MDtvOTkzXFCamnbpWu5rde7sxDfvO9DIG7Xzfh+F7AQAAAACdYPa7AQAA9Mvs+Vk5RUe5bE75ubxymZxyH+aUy9T+LtwuqHi3qNL9kkoPSiovlGUtWFJVOvGdE11t2yCvQyB1p3O03Y7X1THrVgwbXTfodF+truen3kHqkA+rVTyl3sW0VblhiOdqvfpeAAAAAEC/MJIDALChnXrrlM4+c1a5Wzk5RUexREzReFQyJa/iqeJU5Bbd5REcnufpxcsvdqz+Vh2Ow9SZ2o6VcWj16/219tXLrFW2VQKg1f6ga5s0Eradrco10k5Mwwob07Ad792KaatnHjSe7bxrYYWNDQAAAAAMG0ZyAAA2tPThtF74wQvK38tr/v352miOTE75bH55NEfhbkG5bE5O3tE33vyGkjPdX4vj5kuD1wk5yL9ib9aZ3cu6WtUXtp3t1DnomrW/XzFdLzbCPQIAAACA4Xme180KTp8+Lc/z9Morr3SzGnQYzw1AJwzbvyUXn7uod777jqKjUXnV2v8eDdOQa7na/9z+rk9RBQAAAAAAgGCYrgoAgCUnz53UyXMnNXd1Tpm3M5JqIz2SB7o/cgMAAAAAAADBkeQAAGCV5MGkkgdJbAAAAAAAAAw6khwAMEAMw1j+3Gw2wUbnNSvf6tp+6wb8CLKQNOsDAAAAAACAsFh4HAAGRD3JUE8wrEw6+D3P87yGCYpmxwzDWD7ueV7DugEAAAAAAIBBwkgOABgg9SREq0SD3/OC1htE9kpWmStL61YcSis1k2q7HVg/GJ0BAAAAAAB6gSQHAECS/+mqLsxe0PWL1xXbFJMMSUunOkVH+57dp9nzs11uKQAAAAAAAFBDkgMAIOmTa3usTnTcunxLZ46dUWJ7QlNHpxQfi8swDVWdquy8LWve0o03b+jVkVd16q1TSh9O9/oWAAAAAAAAsMGQ5AAAtHTr8i298dU3lJpJaXLvpBI7E4ptismrenLyjkrzJRXvFRUfiyuXzensM2f1wg9e6HezAQAAAAAAsM6R5ACAAVIfQdFqnQ2/5wWtt5Ezx84oNZNSaial7U9u1/iucZkxU07BUelBSSN3RxQdicqMmJKk3K2cXj/6uvQ7HWkeAAAAAAAAsCaSHAAwIOpJi3riotH0Ua3OW/25frzZsdUJk5XXvDB7QYkdCU3undT2J7crOZPUxPSEKnZFhTsFRUejMgxDVbeqil2Ra7lyio6sRUv2BVtiiQ4AAAAAAAB0CUkOABggjUZTrN7v9zy/x5odv37xuqaOTimxM6HxXeOamJ7QlqktsuYtVeyKnKIjO2crPhavbYm4YomYRreOyr5uN60TAAAAAAAAaIfZ7wYAAAZX9kpWsU0xxcfiim2KyYyZqtgVWfOWXMuVV10aVRIxZEZNmTFTZtxUNB5VNB6VopIx15kptQAAAAAAAIDVSHIAwADIZrMPbZL005/+dPn4pUuX+vL5R+d+JBmSYRq1RcYLjgp3CsplcireK8rO23ItV1W3Kq/qSd7SVFimaokPw5QygcMBAAAAAAAA+MJ0VQDQZ6dPn9aePXt0/Pjxh/avnD6qX59rO6SqU5WTry0yHh2Nyik6MgxDruWqvFiWU3DkWq4qTqWW8Kh4taQHAAAAAAAA0EWM5ACAAfDuu+8qnU4vb5J05MiR5eMrEyA9/fx87bOdt1WaL6l4t6h8Nq/chznlsjkV7hZUelBSebEsu2DLKa5KdnievDTJDgAAAAAAAHQHIzkAAA2lZlJyio6seUvFe0VFR6KqulXFx+Iyo6a8qifXcmUXbJUXyrLzS4mOoltbs8P1pGS/7wIAAAAAAADrFUkOAEBT+57dpxtv3qglNiK1hcfjY3GZMVPypIpTkVN0ZOdriY76qA5rwZI+3+/WAwAAAAAAYD0jyQEAaGr2/KxeHXlVuWxOkuRaruKJuMy4KcMwVHWrci1XTtFRebGs8kK5luCoSjopidmqAAAAAAAA0CWsyQEAaOnUW6fk5B3lbuWUy+S0mFlUPpNXLptTfi6vwt2CineLKj0oqfigKM/z9OLlF/vdbAAAAAAAAKxzJDkAAC2lD6f1wg9eUP5eXvPvzyuXqSU78tl8Lclxu6DC3YJy2ZycvKNvvPkNJWdYjAMAAAAAAADdxXRVAABfdh3ZpW+Wv6mLz13UO999R9HRqLxqbS4qwzTkWq72P7dfJ75zos8tBQAAAAAAwEZBkgMAEMjJcyd18txJzV2dU+btjKTaSI/kAUZuAAAAAAAAoLdIcgAAQkkeTCp5kMQGAAAAAAAA+ockBwCgJwzDWP7seV5b5xmG8dCxlWVWl1t9rFX9AAAAAAAAGB4sPA4A6Lp6oqGeXFgr8eD3vEZlPc9b3hrtJ7kBAAAAAACwvjCSAwDQE/UEg+d5DRMVrc6rj+BoVr6Z1SNAGsleySpzZWm9kUNppWZSa543/VqoZujmS63L3nyp9XXWukY3y/nV6N781tGp++p1fd2qs914Nrteq2sEjWm773WvnkWn39FuvWt+/43p5PcXAAAAAIYNSQ4AwFBolaDwOx1WMxdmL+j6xeuKbYpJhqSlyzhFR/ue3afZ87OhrhvG9GvBO71XHlurbNhyQbRbR6PyYePRTKu2drqdYers9DPzG6uwMW1Hp2PaqFw7Me3Wu9asTgAAAABAcyQ5AAADr1WCY/Wxtc5vdo1bl2/pzLEzSmxPaOrolOJjcRmmoapTlZ23Zc1buvHmDb068qpOvXVK6cNpSa07Qv10WjbrhG3V6R2kbNhyYbSKi5/O57Xa3qpt9WNBO6Ibxbhb7VyrztXXbXVu0PrC6lRMWwkb07Dl2o1pkLi0893zk9whOQIAAABgo2NNDgDAUDAMY3mr/90Jty7f0htffUOpmZR2P71bu47sUvpQWskDSe343A5t27NNE7snNLl3UomdCZ195qw+/MmHHam7kbDJET9lw5bzy0/HcNBr+LmnsJ3qQTrAW53ndyqmIM+gE/H0W//qa/ej8zxoTMOUayem7cSl2989AAAAANioSHIAAHrCb3JirfPWWjy8U4uInzl2Rtse36bUTErpQ2k9+sVHlfpCSjue2qGtj2/VxPSENu/arLHkmMZT44o9EtPrR1/vSN34mN8EQK+nUVpd36C2069B7FAPG9NhfxbtWI/3BAAAAABhMV0VAKDr6ouF1xMXKxMUK6eRanZeM6sTJ36nqrowe0GJHQlN7p3U9ie3KzmT1MT0hCp2RYU7BUVHozIMQ1W3qopdkWu5coqOrEVL3/v693Tiz074DwI2tH5PLdTv5NAgJlfWA+IKAAAAAIzkAAD0yOqRGCv3+znPb5m1yjW61vWL17X18a1K7ExofNe4JqYntGVqizZt36TRiVGNbB5RfCz+8ZaIK5aIaXTrqK6dv+b31gNr1iHd6tfpjTrTw5brJH59/rHp1x7ewl4jyHn97BBv5z57qRttHITvHgAAAACsZ4zkAABsSNkrWcU2xRQfiyu2KSYzZqpiV2TNW3ItV151aVRJxJAZNWXGTJlxU9F4tLaNRjV3dU7Jg8m22uEnobHW/madxp0u12mD0Jm7MhbNFnfvtG50dLezhks39DKe3dLpeHXyu0dSBAAAAAAexkgOAENn9QLUQc9rVb7d43jYE088oWw2u7zVrdwXZP/t27d16dKl5f1hP2euZCRDMkxDXtWTU3BUuFNQLpNT8V5Rdt6Wa7mqulV5VU/ylqbFMiUjUiuTeTvToSitf4PcMduJURVB1RewDruQtd949jrB0Grx9EFOeAzyOwoAAAAAaIyRHACGysq1GuqJhrWmImp2Xv2/ayUpVl+v1d9o7uWXX1Ymkwk0hZSf/Z36LE+qOlU5eUelByVFR6Nyio4Mw5BruSovluUUHLmWq4pTqSU8Kl4t6dEhjTpUG40yqB9rVLYb5do1qJ3HjaYR6vaC1Y0SAX6fQdAExyDEvX5/g6rbserXdw8AAAAANgKSHACGzupFqts9b60yayHBEU46ne74/p07dy5/Pn78eKjP6UO169l5W6X5kkbujsgwDNk5W0aktti4U3BUXizLLthyig8nOwzTUPrw2m3thEad3q06Yztdrl2D1NHeyCC3bbUw8WyWXBiG59Nt/Uxw1Pd3OsEFAAAAABsJSQ4AWGVlQmR1UqPZsZU+uvGRfvp7P9V7f/WeHtx4IEna9vg27f6l3TryL45o4rGJDrcaQaVmUnKKjqx5S8V7RUVHoqq6VcXH4jKjpryqJ9dyZRdslRfKsvNLiY6iK9eqbckD7a3Hsd4Nc4fsILZ9ENs07IgpAAAAAAw/1uQAgFU8z1veVo8AaXas7s1/+ab+6PAf6eqZq7LmLW19bKu2fHqLireLuvJHV/RvD/5b/fB3ftiLW0EL+57dV1uD425R+bm8cplcbcvmlJ/Lq3C3oOK9okoflVReKC+P6rAWLO1/bn+/mz/QgnYet5oialA6o8O2s912h7n/1et+rLUGSNg1QYJqNpqk3ZiGfWcG5Z3ya5Cn+wIAAACAfiLJAQAd9KdP/6mu/p9XlTyQ1OP/7HF95lc+o11Hdil5MKnkF5JK/pOkxnaO6T/8H/9Bb/wXb/S7uRve7PlZOUVnOamRy+SU+7CW6MjP5VW4XVDxblGl+yWVHtQSHdaCJVWlE9850dW2terQbNWx2+lyQbTbeby6Ld3q3G03oRK2nWud12rNBj/t6Tc/71avYur3e9DLmHbquzfo7wEAAAAA9BrTVQEYOvW1MVqts+H3vLXKhHH2mbOa+9s5TR+f1uSeSY2lxhQdjapiV1ReKKt4v6jieFGR0YiMqKGbb93UG7/6hnQ4VHXokFNvndLZZ84qdysnp+gologpGo9KpuRVPFWcityiuzyCw/M8vXj5xY7V36qDc61fr9fL+Pl1fLvlglp57aD1tGpjq1/kt9rfLIEQRNh2+n0GjdrXzefWrN5m+1fXGzYh1a2YtnrmQWMaJi69+u4BAAAAwEZFkgPAUKknLeqJi5UJiZUJilbnrf7caJHyleWaHftPF/+Tbv34lqZ+cUqf+vyntHP/Tm1+dLNkSNa8pcKdgqKjURmmIa/qqepUVbErev9H70ubJe1tNzIIK304rRd+8IJeP/q6rEVLo1tHFY1HZUSWnpVblWu5yyM4Xrz8opIz3V+Lo1mHZ7NperpRrpdWdgiv3j9IdYUt28v767VG91Y/Frb8eohpu9+9YRnNAwAAAAD9YHhhf7Ls0+nTp+V5nl555ZVuVoMO47kBwfz+U7+vSCyiR3/hUT36nz2q9JG0JvdOqnS/9PE6D6u3D3NavLWoggry/ju+b4Pg4nMX9c5331F0NCqvWvvfo2Eaci1X+5/b3/UpqgAAAAAAABAMIzkAoE35bF4P/vGBpo9O65Gtj2hkYkTxRFySZEZNRUYiio5GP7k9ElUsEZM+kIyC/ym10D0nz53UyXMnNXd1Tpm3M5JqIz2SB7o/cgMAAAAAAADBkeQAgDZlrmSWkxlGxFDVrcqatyRDy1MdVd2q5NWmxzJMQ2bElBmtbYZpyMt0dVAdAkoeTCp5kMQGAAAAAADAoDP73QAA8OPSpUu6cuXKQ1vduXPnlj+fPn2655/n/3FeZsSUV/VUKdcWGc/P5ZXL5FS8U1R5viyn4Mi1XFWciryKV5sKaSnpETEi0oPgMQF6Yfo1/xsAAAAAAECvsSYH1sRzw6AZ5Hfy3f/7Xf27X/t32nVkl7Y/uV0Tuyc0tnNM8fG4zIipil2RnbdlzVsq3i+qeLeowt2CCrcLys/lNf/BvLxnPb38nZf7fSvAJwRJXrAoMgAAAAAA6DWmqwKANqW/kFa1UlV5sazSg5Jim2Lyqp7iubjMqCmv4skpObJzdm0r2HKLbm1kh12RV/XkpZmuCoOJxAUAAAAAABhkJDkAoE1jqTFNfnZSpXslFbYUZEZNVZyK4mNxRWKR2jRWdkV2wZa9uLTlbTlFR07JkbZLSvT7LgAAAAAAAIDhQ5IDADrgl//nX9bF5y8qNh6TYRhyy+5ykkOSqk5VjuXIztsqL5ZlLVqy87bckit9rc+NBwAAAAAAAIYUSQ4A6IAn/qsn9Nixx3Tzr2/Kq9amp4on4orEI5IheZXaaA6nuJToWCireL+oz/7nn9XP9vxMYrYqAAAAAAAAIDCz3w0AgPXi+X//vKa+MqWFny8on8krl8kpl8kpn80rfzuvwu2CCncLKt4rqnCvoM/+ymf19e9/vd/NBgAAAAAAAIYWIzkAoINe+PMX9MN/9UO9/ftvK387r8hIRNGR2j+1ruWqUq4o8khEv/Avf0HHTx/vb2MBAAAAAACAIUeSAwA67Ff+9a/o0H97SD/9/Z/qvR++p/t/f1+StG3vNn3mVz6jI795RJsf3dznVgIAAAAAAADDjyQHAHTBluktevp/fbrfzQAAAAAAAADWNdbkAAAAAAAAAAAAQ4kkBwAAAAAAAAAAGEokOQAAAAAAAAAAwFAiyQEAAAAAAAAAAIYSSQ4AAAAAAAAAADCUSHIAAAAAAAAAAIChRJIDAAAAAAAAAAAMJZIcAAAAAAAAAABgKJHkAAAAAAAAAAAAQ4kkBwAAAAAAAAAAGEokOQAAAAAAAAAAwFCK9rsBAICPGYax/NnzvLbOMwzjoWOtyvitGwAAAAAAABgUjOQAgAFRTzLUEwwrkw5Bz1trn+d5LRMi9a1R3QAAAAAAAMAgYSQHAAyQehKiVaKh2Xn1hEWQREWYkRvZK1llrmQkSelDaaVmUmueN/1a4EtLkm6+1LrszZdaX2etazQq57etfuptpVFdfq8d5L6alet1fd2qs914Nrteq2sEjWm773WvnkWn39Fuv2udKg8AAAAAGC4kOQBgHVk9RVXQsnXNrnFh9oKuX7yu2KaYZEhaOtUpOtr37D7Nnp8NVX8Y06+FS1jUj/Wr47PdtjUqHzYezbRqa6fbGabOTj9rv7EKG9N2dDqmjcq1E9NuvWvN6vRTnkQHAAAAAKxPJDkAYJ1oJ8Eh6RPrd6y+1q3Lt3Tm2Bkltic0dXRK8bG4DNNQ1anKztuy5i3dePOGXh15VafeOqX04bSk1h2hfjoem3XCtur0DlLWTwdqpzpKW8XFT+fzyuOtyq0uE7QjulGMu9XOtepcfd1W5watL6xOxbSVsDENW67dmAaJS9jvbLN6G10fAAAAALC+sCYHsE4ZhrG8hTnPT/m1zvFbL7pjdfw79RxuXb6lN776hlIzKe1+erd2Hdml9KG0kgeS2vG5Hdq2Z5smdk9ocu+kEjsTOvvMWX34kw87UncjYZMjfst2k5+O4aDX8NPxG7ZTPUgHeKvz/E7FFOTZdSKefutffe1+vEtBYxqmXDsxbScuYb+zYd4bAAAAAMD6QJIDWIc6sYB1s0Wq6+euXKjabzk05zc5sdZ5K5/HyjU7gtTbyJljZ7Tt8W1KzaSUPpTWo198VKkvpLTjqR3a+vhWTd2z4jwAACAASURBVExPaPOuzRpLjmk8Na7YIzG9fvR1X3UPg0H5Fbjfjtxet3d1fYPaTr8GsUM8bEyH/VkAAAAAANAK01UB61QnFrBupN1pkdZy/937kqTJvZMdve4wqT+D1ckn6eGYNzuvmdUjblaWXf38V17zwuwFJXYkNLl3Utuf3K7kTFIT0xOq2BUV7hQUHY3KMAxV3aoqdkWu5copOrIWLX3v69/TiT87ESYcA2kQO7/Xk0FeK6UX9fF+hdPv9wYAAAAA0F8kOQCE4neR6mau/vFVXf7WZT34hweKxmv/HLm2q8nHJ/Xl3/6yDpw60JG2DpNGsVy930/Mg5ZpdPz6xeuaOjqlxM6ExneNa2J6Qlumtsiat1SxK3KKjuycrfhYvLYl4oolYhrdOqpr5691LcnRrEP65ku1463WKBjETlF+Uf+xTiQBgi4i3s93YhDa4Ec33tFOfmdJHgEAAADAxkKSA0AorRapbsYtu/qTI3+ihVsLemTrI9p1aJfMuLm8gHXhbkF/8d//hX7yez/Rb/zkN2REWN+jX7JXsoptiik+FldsU0xmzFTFrsiat+Rarrzq0qiSiCEzasqMmTLjpqLxaG0bjWru6pySB5NttaNVQqPR/nqnaZByjerudUfpIHTMNut47mYyptG123kW7azh0g29jGe3dDpe7X5nu/HeAAAAAAAGH0kOAD1VzpX1e0/8niLxiKa/Mq2x5JhiiZi8qie35Mqat5TYkVDhXkEfvfeRvjX1Lf3mu7/Z72Z33RNPPKFsNrv8dyqVkqSH9vnZf/v2bf3d3/2djh07Jkm6dOlSW5/HfjYmGZJhGvKqnpyCo8Kdgip2RV7Vk5235Vquqm5VXtWTvKVRPqZkRGplMm9n2k5ybBSD3Bnbj074dhMBfuPZ63trtHh6vR2NRjMMgkF+R+vWQwIJAAAAAOAfSQ5gnaqPrvCzgHWQ9Tja9Qef/wPFEjE9+sVHNfnEpMbT44rEI3KtWoKjeK9YGzEQN2UYhhY+WNAf7v9D6Rs9aV5fvPzyy8pkMmuOhvE7fVWjYx357ElVpyon76j0oKToaFRO0ZFhGHItV+XFspyCI9dyVXEqtYRHxaslPTqkUYdq2OltmpXrl0HtPG60MHW3F6xulgjw8+yCJjgGIe4rEx2DqNux6sR3tt33BgAAAAAwfEhyAOtQJxawDrtIdbNyf/k//qXKi2V95unPaMe+HUr+k6Q2f3qzvKqn0v2S8nfyMqNmrVO9voh12dXCzQXpR5KOtR2agZVOpzuyf+fOndq5c+fy38ePH2/rc3ZzbcSInbdVmi9p5O6IDMOQnbNlRGqLjTsFR+XFsuyCLaf4cLLDMA2lD6/d1k5o1HnZqjPWb6dnrzrAB6mjvZFBbttqYeLZLLkwDM+n2/qZ4KjvJ1EBAAAAAFgLSQ5gnWp3Aeuwi1Q32l91q3r7D9/W5N7J5cWrJ5+Y1NbPbFXpQUmGadSSGkVXTqG2kHU5V1Z8LK6RLSNyfuLI+EXW5ui11ExKTtFZHmUTHYmq6lYVH4vLjJq1acYsV3bBVnmhLDu/lOgounKt2pY8wFRVzQxzB/ogtn0Q2zTsiCkAAAAAYJCZ/W4AgI3ho3/8SG7R1ejWUY2Mjyi2KSbDMOQUndri1aZRW7Q6ZioSjygyElF0JKroSFSxR2KSI3kfdW76I/i379l9ymVyKt4tKj+XVy6Tq23ZnPJzeRXuFlS8V1Tpo5LKC+XlUR3WgqX9z+3vd/ND68W0QUE7j1tNETUondFh29luu8Pc/82XGm+rz+m2Zu9cuzEN+84MyjvVzCC3DQAAAADQfSQ5gHUml8s9tK3cf+fOneW/b9682dPP2StZyZAisYgkyS27Kt4vKpfJqXS/JKfgLC9m7Xne8mLXRsSQGTVlGqaUCR4PtG/2/KycorOc1Mhlcsp9WEt05OfyKtwuqHi3qNL9kkoPaokOa8GSqtKJ75zoattaJSJadez60a/pefyWb/R3p7SbUAnbzrXOa7Vmg5/29Jufd7JXMfX7/ellTNv9zgZ9bwAAAAAAw4/pqoB15Nvf/rZ2796tZPLj6YHGx8clSdlsVvfu3dOnPvUpSdJf/dVf6dd//dd79vmp3FMyzKU1HEqOrI8s5efyywtYO6XaFFVuyVXFrqjqVFWtVKVqbQos0zBVLVY7HjP4c+qtUzr7zFnlbuXkFB3FEjFF41HJlLyKp4pTm2qsPoLD8zy9ePnFjtXfqoNzrV+v18v4+XV8r61sU9D2tbq3Vr/Ib7W/WQIhiLDt9PvsGrWvl887bEzDJqS6FdNWzzxoTMPEpd3vbJj3BgAAAACwPpDkANaR999/X++9955eeeWVTxzbu3ev9u7du/x3PQHRq883L92UV/VqazfMl1W4W5BhGrLztsyIqYpdqR1bLMspLC1ebVdUcSryXE+VakXeTqar6pf04bRe+MELev3o67IWLY1uHVU0HpURMeRVPVXdqlzLXR7B8eLlF5Wc6f5aHM06rZtN0+P3l/KD+svvlR26q/cPUl1hy/by/nqt0b3Vj4Utvx5i2s53tn7OoN8jAAAAAKDzSHIA6InUF1Kq2BWVF8oq3i8q+khUXtVTPLe0gHWltoB1Obe0pkN9AWvLlVt25VU8GbtYeLyfdh3ZpW+Wv6mLz13UO999R9HR2jOUalOLuZar/c/t9z1Fld9Oy3aFuUa3O0X7cV/t1NmPskHX1egGP4mDbly3W+V7FdN+3V8n6gYAAAAADB+SHAB6Ij4W16e/9Gnd//v7GhkfkRk1VXEqiifiMmOm5EkVuyKn6NQSHQtl2TlbdsGWnbelKcmLMpJjEJw8d1Inz53U3NU5Zd6uLZSSPpxW8kD3R24AAAAAAAAAK5HkANAzXzv7Nf2bvf9G+Tt5yagtPh5PxBWJ1xYjr095VJ+2qp7ocC1XOtnnxuMTkgeTSh4ksQEAAAAAAID+IckBoGe2TG3RL778i3rrf3pLXtWTU3I+TnIYSwtY25XaIuT5pURHrqxf/te/rDcLb0oM5MCACrLQMdPpAAAAAAAAdA5JDgA9dfR/OKrYaEx/+a/+UnbO1sj4iCIjERmmIc/zVHWWRnPkbDmWo3/6v/1THf7Nw3rz9Jv9bjoAAAAAAACAAUOSA0DPffG3vqipX5jShV+7oPxcXhW3okisNmVVxa4oEo8osT2hf/7//HMlZ5gOCYOP0RkAAAAAAAD9QZIDQF+kj6T10j++pJ//zc/13g/f0weXP5Akfforn9buX9qtqa9M9bmFAAAAAAAAAAYdSQ4A/WNIU1+ZIqEBAAAAAAAAIBSz3w0AAAAAAAAAAAAIgyQHAAAAAAAAAAAYSiQ5AAAAAAAAAADAUCLJAQAAAAAAAAAAhhJJDgAAAAAAAAAAMJRIcgAAAAAAAAAAgKFEkgMAAAAAAAAAAAwlkhwAAAAAAAAAAGAokeQAAAAAAAAAAABDiSQHAAAAAAAAAAAYSiQ5AAAAAAAAAADAUCLJAQAAAAAAAAAAhhJJDgAAAAAAAAAAMJRIcgAAAAAAAAAAgKFEkgMAAAAAAAAAAAwlkhwAAAAAAAAAAGAoRfvdAAAIyjCM5c+e54U6r9GxlfubXd8wjKZ1AwAAAAAAAOg+RnIAGCr1JEQ9wbA6KeHnvHqCor6tldiob82uDQAAAAAAAKC/GMkBYOjUkw9rJSj8nNfOCIx6gsRvoiN7JavMlYwkKX0ordRMas3zpl8L156bL7Uue/OlxscalW1WplX5XpQNc+0g1w/bttXlel1fN+tsN6aNrtWqfNCYtvN9aFS+G8+i0+9o2Db6uUYnnz0AAAAAAJ1GkgPAhhR2Kiu/CZILsxd0/eJ1xTbFJEPSUjGn6Gjfs/s0e342fOMDmn5t7c7IZh2e9WNBOz07UbbdjtN22tasfLO2hU1StROLMO0MW2e7MfV7rTDndVKnY9rr7143dPLZAwAAAADQDSQ5AGxIjZIXayU86iM3/CQ4bl2+pTPHziixPaGpo1OKj8VlmIaqTlV23pY1b+nGmzf06sirOvXWKaUPpyW17tD004nYrDO1WSdt0HKtfoXvp861yvot50erePrpfF55POg9Be2IDhqLdtu5Vp2rr+vn/KB1htGpmLYSNqZhy7Ubz7BxaVZ30PO7/ewBAAAAAPCDNTkAwCfDMJa3+t8r3bp8S2989Q2lZlLa/fRu7TqyS+lDaSUPJLXjczu0bc82Teye0OTeSSV2JnT2mbP68CcfdrXNrTpJgyQ+/J7j5xfvQTp9gwqSWPF7np+EQdhO9XZiEbSdrZJmYZ5pkPsOmrTrR8d50JiGKddOPHsdl049ewAAAAAAuoUkB4Ch0yjJ4Pe8MAuHr1yMvNGojzPHzmjb49uUmkkpfSitR7/4qFJfSGnHUzu09fGtmpie0OZdmzWWHNN4alyxR2J6/ejrgduyEfRzqqJWHf39ni5oUNvp1yB2jIeN6bA/CwAAAAAA1gOmqwIwVOpTR9UTFc2mnWp0XrOFyFcnQIKswZHYkdDk3kltf3K7kjNJTUxPqGJXVLhTUHQ0KsMwVHWrqtgVuZYrp+jIWrT0va9/Tyf+7ETASADB9Hv9hH4nhwYxudIvxAYAAAAAsJ4wkgPA0Fk9omLlfj/ntbpGs3KN6rp+8bq2Pr5ViZ0Jje8a18T0hLZMbdGm7Zs0OjGqkc0jio/FP94SccUSMY1uHdW189eChsC3dhfEbjaVUrPFlhuV7Td+Uf+x6dce3tq5jt9z+vlOtHufvdKvUUxh6h2GeAIAAAAA1j9GcgBAm7JXsoptiik+FldsU0xmzFTFrsiat+Rarrzq0qiSiCEzasqMmTLjpqLxaG0bjWru6pySB5NttSPMotHNyraai79Zx2iz6Xvq5ZotaN5tg5CA6VcsupWcCrPQebf0893qlG7FqxuxGYTvEwAAAABg42IkB4Ch8cQTTyibzS5vdSv3Ndt/5cqV5WOXLl3q2OfMlYxkSIZpyKt6cgqOCncKymVyKt4rys7bci1XVbcqr+pJ3tK0WKZkRGplMm9nOhChzup2x3CnRhIEqU8azA7ZXsdC+ngB63YWsvYT014nGFot5D7ICY9uv6OdjM0gf58AAAAAABsLIzkADIWXX35ZmUym4dRTa2k2nVXHP3tS1anKyTsqPSgpOhqVU3RkGIZcy1V5sSyn4Mi1XFWcSi3hUfFqSY8OadTZ2Gi0QKuyzco16+BsVV+j6a66uUjzoHbI9iMWK6+/ep+fd6UuSIJjEOJev79B1c9YBY3NID1XAAAAAABIcgAYGul0uq39K/8+fvx4xz5nN9dGj9h5W6X5kkbujsgwDNk5W0aktti4U3BUXizLLthyig8nOwzTUPrw2vfQCUE7r1uVa9XB6be+XnWQDkOH7CC3bS1BY9qsA30Ynk+3DVMMhqmtAAAAAICNgSQHALQpNZOSU3RkzVsq3isqOhJV1a0qPhaXGTXlVT25liu7YKu8UJadX0p0FF25Vm1LHmhvPY71otMdqMPcITuobR/Udg2rYYrnMLUVAAAAALBxsCYHAHTAvmf31dbguFtUfi6vXCZX27I55efyKtwtqHivqNJHJZUXysujOqwFS/uf29/v5q9LQTtkW00RNSgdvGHb2Yl2h4lpo231Od3WbDRJuzEN+84MyjvlZ6qqQWkrAAAAAACrkeQAgA6YPT8rp+gsJzVymZxyH9YSHfm5vAq3CyreLap0v6TSg1qiw1qwpKp04jsnutq2Vh2wQct167qd7ERt91qr29ittRzajUXYdq513rB0yDfj553sVUz9fg96PW1bs/3D/OwBAAAAABsX01UBQIeceuuUzj5zVrlbOTlFR7FETNF4VDIlr+Kp4lTkFt3lERye5+nFyy92rP5WnarNFhAPUm7lIsV+fh2/lm4uAL3y2kHb1+reWnUCt9of9hms1Y4w7fT77FZrJ6ZhhY1p2HerWzFt9cyDxrPX71o/nj0AAAAAAEEwkgMAOiR9OK0XfvCC8vfymn9/fnnKqnw2vzyao3C3oFw2Jyfv6BtvfkPJme6vxdFoOqBWnZLNphEKe6xZvb2atqiVZu3rZV1+nk+Q/Z2oc9C1eu/6EdNB0W5sAAAAAAAYVIbneV43Kzh9+rQ8z9Mrr7zSzWrQYTy34cRzGxwXn7uod777jqKjUXnV2j+zhmnItVztf25/16eoAgAAAAAAADYCpqsCgC44ee6kTp47qbmrc8q8nZFUG+mRPND9kRsAAAAAAADARkGSAwC6KHkwqeRBEhsAAAAAAABAN5DkAAAMtCCLJbOuAAAAAAAAwMbCwuMAAAAAAAAAAGAoMZIDADDQGJ0BAAAAAACARhjJAQAAAAAAAAAAhhJJDgAAAAAAAAAAMJRIcgAAAAAAAAAAgKFEkgMAAAAAAAAAAAwlkhwAAAAAAAAAAGAokeQAAAAAAAAAAABDiSQHAAAAAAAAAAAYSiQ5AAAAAAAAAADAUCLJAQAAAAAAAAAAhhJJDgAAAAAAAAAAMJRIcgAAAAAAAAAAgKFEkgMAAAAAAAAAAAwlkhwAAAAAAAAAAGAokeQAAAAAAAAAAABDiSQHAAAAAAAAAAAYSiQ5AAAAAAAAAADAUCLJAQAAAAAAAAAAhhJJDgAAAAAAAAAAMJSi/W4AAGBjMAxj+bPneW2dZxjGJ441K+e3bgAAAAAAAAwXRnIAALqunmSoJxhWJh2Cntdon+d5y9vKc5odAwAAAAAAwHBjJAcAoCfqiYtWiYZm59UTFkESFWFGbmSvZJW5kpEkpQ+llZpJrXne9GuBLy1JuvlS67I3X2p8rFHZZmValQ9b1m+5MNcOWkfY+wp7T52qr5t1thvTRtdqVT5oTNv5PjQq341n0el3NGwbw9S9+lqd/O4CAAAAQD+R5AAADIW1pqiqW534CDtd1YXZC7p+8bpim2KSIWnpVKfoaN+z+zR7fjb8DQQ0/dranZDNOjzrxxp1XrYqG6Zzt1Paua9m5ZvdV9h76kYc24l/N96VINcKc14ndTqmvf7u9cogtAEAAAAAuoEkBwBg4DVLcKx1fPXfzY5J0q3Lt3Tm2Bkltic0dXRK8bG4DNNQ1anKztuy5i3dePOGXh15VafeOqX04bSk1h2afjqQm3WmNuukDVturbJ+y60s240O01bx9NP5vPJ40HgEvaegcWy3nWvVufq6fs4PWmcYnYppK2FjGrZcu/Fs5/vDyAsAAAAAWBtrcgAAhoJhGMtb/e9OuHX5lt746htKzaS0++nd2nVkl9KH0koeSGrH53Zo255tmtg9ocm9k0rsTOjsM2f14U8+7EjdjbTqJA2S+Khrlnjxm4zpZidrkKSM3/P8JAzCdqqHjeNa5/mdiinoc+9ETP3Uv/q6/eiMDxrTMOXaiWe/4lIXdOQOAAAAAAwTkhwAgJ7wm5xY67yVC4evXLMjSL2NnDl2Rtse36bUTErpQ2k9+sVHlfpCSjue2qGtj2/VxPSENu/arLHkmMZT44o9EtPrR1/3VfcwGrYpbfwmAHp9X6vrG9R2+jWIneNhYzrszyKsQXyGAAAAANAJTFcFAOi6+poZ9YRDo+mjmp3n5/or//Zz7MLsBSV2JDS5d1Lbn9yu5ExSE9MTqtgVFe4UFB2NyjAMVd2qKnZFruXKKTqyFi197+vf04k/OxEiGoB//f4Ffr+TQ3TMfyxsbNZbsgYAAAAAViPJAQDoiWaLhvs5r9U5zco1Onb94nVNHZ1SYmdC47vGNTE9oS1TW2TNW6rYFTlFR3bOVnwsXtsSccUSMY1uHdW189e6luRod0Hs9dYxTCftxzqVBPAT00F4nwahDX70c8H1sNPUAQAAAMB6QZIDALAhZa9kFdsUU3wsrtimmMyYqYpdkTVvybVcedWlUSURQ2bUlBkzZcZNRePR2jYa1dzVOSUPJttqR5hFo5uVbTYFz/Rray+MPCxJhEHoqO1XHBtdu91O7DALnXfLsL6XK3UrXu3EZhC+NwAAAADQTSQ5AABdlc1mH/o7lUp1ZP/777+vL33pS5KkS5cu6dixY4E+j/1sTDIkwzTkVT05BUeFOwVV7Iq8qic7b8u1XFXdqryqJ3lL63uYkhGplcm8nWk7ydFpa3W+r3XOsBjkX6L3I46dSAQEGQHQK40Wcq+3w8973S/dfkfDxmaYvucAAAAA0A6SHACArjl9+rT27Nmj48ePf+KY3+mrGu1fve5GqM+eVHWqcvKOSg9Kio5G5RQdGYYh13JVXizLKThyLVcVp1JLeFS8WtKjQxp1jDYaLdCqbLNyjRZUHtSFlgc1wdGvODbr7PabBBi2KY5WduYPon7GqllsBukZAgAAAEC3keQAAHTVu+++q+eff/4T+9Pp9Jrn+92/8u+VSRS/n7ObayND7Lyt0nxJI3dHZBiG7JwtI1JbbNwpOCovlmUXbDnFh5MdhmkofXjttnZC0M7rIOWGoeNzGDppB7ltawka02bJhWF4Pt02DDHgGQIAAADYCEhyAAA2pNRMSk7RkTVvqXivqOhIVFW3qvhYXGbUlFf15Fqu7IKt8kJZdn4p0VF05Vq1LXlgsKaqascgdXgOUluCGtS2D2q7hhXxBAAAAIDBQZIDG5phGMufG02R0+w8P+XbKQugu/Y9u0833rxRS2xEaguPx8fiMmOm5EkVpyKn6MjO1xId9VEd1oKl/c/t73fz16WgncetRq4MSmd02HZ2YrqmMDHt1LXa1eze241p2HdmUN6pVrFpVa7f7QcAAACATjH73QCgX+pJhnqCYWXSwe95nue1TI7Uz1md4Fi5v1HdALpr9vysnKKjXDan/FxeuUxOuQ9zymVqfxduF1S8W1TpfkmlByWVF8qyFiypKp34zomutq3VXPtBy/m5Zr87Pdttx+r769ZaDu3GMWw71zpvWDrkm/Hzrvcqpn6/X/1O8gSJDQAAAACsd4zkwIZWTzy0SjT4PW+leiIDwGA79dYpnX3mrHK3cnKKjmKJmKLxqGRKXsVTxanILbrLIzg8z9OLl1/sWP2tOlWb/dI8aDm/ZYOUa7T4djvXD/oL9ZWjHdYqGzQWfu4pTBzDtrNVuUbaiWlYYWMa9r3sVkxbPfOg8ez1uwYAAAAAGwlJDqCLGk1JtTpZ0iwZMvf/zenH//uP9cHffKCFny9IkrZMb9HUL0zpS7/1Je08sLMLLQc2jvThtF74wQt6/ejrshYtjW4dVTQelREx5FU9Vd2qXMtdHsHx4uUXlZzp/locfjpng5RrVnY9/BK8l/fWTl1hy27EZ1c/Frb8eohpu7EBAAAAgI3A8Lr8U/PTp0/L8zy98sor3awGHbYRntvqkRaNRl74Oc/PvpV/+637+6e+r599/2cyTEOPbHtEo5tHVXWrKj0oqbRQkmmaemr2Kf3qH/2qpI3x3DBchu2dvPjcRb3z3XcUHY3Kqy59X01DruVq/3P7uz5FFQAAAAAAAIJhJAcwoP748B9r/v15pWZSSuxMKD4Wlzwt/6LcemCpeK+oa+eu6fa123rxx52bPgfYqE6eO6mT505q7uqcMm9nJNVGeiQPdH/kBgAAAAAAAIIjyYENrT6CotU6G37P65QzXzmjhQ8W9Njxx7RtzzaNp8YVHY3KLbsqL5RVvF9UYaygyEhERsTQnXfu6Nu/9G3peE+aB6x7yYNJJQ+S2AAAAAAAABh0JDmwYdWTFvXERaPpo1qdt/pzo0XK/a7J8R//r/+o29du67Fjj+lTn/+UPrX/U9r86GZJkjVvqXBnKblhLK0X4FRVsSv68KcfSpOSPt9eXNBfftdqaXZeo2Ork3SNrt9o+jQgiCCLJbO2AAAAAAAACIskBza0Rh25q/f7PS/I8UbH/vp3/1rbPrtNm6c2a2L3hHbs26HtT2xX8W5RuUxOXsVTpVyRW3LlFB3ZeVvxfFwjYyNyfuSQ5BhiKxNl9cRao3ViGp3X6hqt3tlejVYCAAAAAAAAOoEkBzBAFm8tavHWoqaOTml0YlSjW0YVeySmqlOVETFkxk1FRiIPbdHRqGKjMUU3RaUPJOX6fRdoR6ORQEHO83uN1YJOy5a9klXmytK6FYfSSs2kfNeF9Y/RGQAAAAAAoBdIcmBDqlQqD/0diUSW9xuGIcdxNDIyIkkql8s9+/zz//fnMiKGIvGIDNNQxamo9KAkeVLVrcotuao6VcmrdUgbpiEzYsqIGopEIzJNU5XMw/cGrNRsKiu/U1RdmL2g6xevK7YpJhmSloo5RUf7nt2n2fOznWwyAAAAAAAA0JDZ7wYAvfYXf/EXunr1qq5du7a81V27dk03b97Ut771reV9vfz8/W9/X2bElFf15FqurAVL+dt55TI5Fe4UZM1bsgu2XMtVxa7Iq3jyql6tk9mQTMOUMc90Q1ib53kPbfWEh98Ex63Lt/S7sd/VB3/zgaaOTmn62LQeO/6Ypr4ypeSBpHY8tUM33ryhV0deVeanmW7fDgAAAAAAACDD6/LqsqdPn5bneXrllVe6WQ06bD0/t0G+t3/483/Q+dnz2nV4lyafnNTEYxMaS45pZHykNrLDrsgu2LLmLZXul1S8V1ThTkGF2wXlb+c1/8G8vF/z9PKfvtzvW0EIq5MNzdbkaHSe32usPNZoeqqV5W5dvqU3vvqGtj2+TZN7J5XYmVBsU0xe1ZOTd1Sar72PxbtF5bI5OXlHL/zgBf3Jn//JwH7fAAAAAAAAMPyYrgoYIOlDaVXdqsqLZZXul5Y7ke1xW0bEkFfx5JRqi42Xc2XZBVtOyZFbXhrZUfWkdL/vAu3wuy5Gs/OCrq2xOgmyVmLkzLEzSs2klJpJafuT2zW+a1xmzJRTcFR6UNLI3RFFR6IyI7UBgrlbOb1+9HXpd3w1AQAAAAAAAAiFJAcwQDbt2KQdn9uh4r2i4pvjMmOmqk4t6RGJReR5nirlipyio/JiWeWFsuy8LafgyC7Y0qck75GuDs5CF9UTE/XkzjGs8wAAIABJREFURKMRGc3Oa3WN1fX5cWH2ghI7EprcO6ntT25XciapiekJVeyKCncKio5GZRiGqm5VFbsi13LlFB1Zi5bsC7bEEh0AAAAAAADoEpIcwIB55lvP6NxXzyk+FpdhGKqUK4olYorEaoujV92qXMutjeZYSnSUc2VVyhXpa31uPNrWKPGwen+zBIXfa/gtf/3idU0dnVJiZ0Lju8Y1MT2hLVNbZM1bqti1pJudsxUfi9e2RFyxREyjW0dlX7d91QkAAAAAAACEQZIDGDCP/dJj2vNf7tG7//7d5QXIY4mYIvFI7dfylWptNEep1rFcXiyrNF/SU197Sn879be1RciBDsleySq2Kab4WFyxTTGZMVMVuyJr3pJrufKqSyNHIobMqCkzZsqMm4rGo4rGo1JUMub8TZsFAAAAAAAABGX2uwEAPmn2u7N68r9+Uou3FpXL5JT7MFf7bzan/FxehTsFFe8WVbxfVOlBSZ9/9vP62lmGcQyzbDarbDarv//7v1/ed+nSpb5//tG5H0mGZJhGbZHxgqPCnYJymZyK94qy87Zcy1XVrdbWhPGWpsUyVUt8GKaUCRQKAAAAAAAAwDdGcgAD6sR3TujH3/qxLr1ySYU7BZmx2q/jPdXW5ai6VUViET39vzytI//iSL+bizacPn1ae/bs0fHjxz+xvka/P9d2SFWnKidfW2Q8OhqVU3RkGIZcy1V5sSyn4Mi1XFWc2rvpVbxa0gMAAAAAAADoIpIcwAD70m99STP/zYyu/MkV3fjBDd25fkeSlDqY0uP/7HHN/MaMRjaP9LmV6IR3331Xzz///EP7jh8/3v/Pzx/Xe3/wnuy8rdJ8SSN3R2QYhuycLSNSW2zcKTgqL5ZlF2w5xVXJDs+TlybZAQAAAAAAgO4gyQEMuJGJEX35t7+sL//2l/vdFGxAqZmUnKIja95S8V5R0ZGoqm5V8bG4zKi5vG6MXbBVXijLzi8lOopubc0O15OS/b4LAAAAAAAArFckOQAATe17dp9uvHmjltiI1BYej4/FZcZMyZMqTkVO0ZGdryU66qM6rAVL+ny/Ww8AAAAAAID1jCQHAKCp2fOzenXkVeWyOUmSa7mKJ+Iy46YMozZllWu5coq1aavKC+VagqMq6aQkZqsCAAAAAABAl5j9bgAAYPCdeuuUnLyj3K2ccpmcFjOLymfyymVzys/lVbhbUPFuUaUHJRUfFOV5nl68/GK/mw0AAAAAAIB1jiQHAKCl9OG0XvjBC8rfy2v+/XnlMrVkRz6bryU5bhdUuFtQLpuTk3f0jTe/oeQMi3EAAAAAAACgu5iuCgDgy64ju/TN8jd18bmLeue77yg6GpVXrc1FZZiGXMvV/uf268R3TvS5pQAAAAAAANgoSHIAAAI5ee6kTp47qbmrc8q8nZFUG+mRPMDIDQAAAAAAAPQWSQ4AQCjJg0klD5LYAAAAAAAAQP+Q5AAA9IRhGMufPc8LdV43jgEAAAAAAGB4sfA4AKDr6kmGeoJhZdLB73mGYcjzvOWtE8cAAAAAAAAw3BjJAQDoiXriolWiodF5zUZghD3WSPZKVpkrS+uNHEorNZNa87zp1wJfWpJ086XWZW++FOyaK6/XqGyjOoPUtdY1grY1yLWDXD9s21aX63V93ayzE898rWu1Kh80pu1+H3r1LDr9joZtY5A6G12jU99bAAAAAOg3khwAgKHR7SmpLsxe0PWL1xXbFJMMSUuXcYqO9j27T7PnZ0NdN4zp19rr+A5yTv1Y2I7oIG0Neu2Vx8J0PjdrW9gkVau2drqdYevsxDP3c60w53VSp2PajXh2611rVqef8iQ6AAAAAKwHJDkAAENjZfKiPg2Vn2Ot9t+6fEtnjp1RYntCU0enFB+LyzANVZ2q7Lwta97SjTdv6NWRV3XqrVNKH05Lat2h6acDsVlnajc6IcPW1+hX/J1sa6u6/XQ+h2lb/VjQjujV1+x2O9eqc/V1/ZwftM4wOhXTVsLGNGy5duMZJi6deIZB3xsAAAAAGCasyQEA2BCaJTje+OobSs2ktPvp3dp1ZJfSh9JKHkhqx+d2aNuebZrYPaHJvZNK7Ezo7DNn9eFPPuxqW8NMQ+Onk7PROUHqW31upzrI/XQMB71GOzFppFm8u9XOVs84zHPtxjtWP6cfowPCvpdByrUTz7BxaafOsO8NAAAAAAwbkhwAgJ6oTyfVauHvRuc1K+fnmo2msDpz7Iy2Pb5NqZmU0ofSevSLjyr1hZR2PLVDWx/fqonpCW3etVljyTGNp8YVeySm14++3rS+Xut2Z6XfztJ+TlU0aG1bXd+gttOvQewQDxvTYX8WAAAAAICHMV0VAKDr6ouI15MRjaaWanZes4XImx1bK2lSP35h9oISOxKa3Dup7U9uV3ImqYnpCVXsigp3CoqORmUYhqpuVRW7Itdy5RQdWYuWvvf17+nEn51oPzhtoBN2/Qu6dka36u9XfYOYXBkG/X5vAAAAAKCXSHIAAHqi0UiK1fubLRoe5lizMtcvXtfU0SkldiY0vmtcE9MT2jK1Rda8pYpdkVN0ZOdsxcfitS0RVywR0+jWUV07f61rSY4gC4l3ohNz0DtESeZ8rFNJgF6/Y2ENQhv8GKQF1/2cO+jxBAAAAIAgSHIAADak7JWsYptiio/FFdsUkxkzVbErsuYtuZYrr7o0qiRiyIyaMmOmzLipaDxa20ajmrs6p+TBZFvtCLNotN/jQeochk7PQWjjzZdq8VtrsedudnQ3una7SYBuvWNh9DKe3TII65Gs1K33BgAAAAAGCWtyABg69emM/KzDsNZ5rco3O+63bnzsiSeeUDabXd7qVu5rtP/tt99e3n/p0qWOfs5cyUj/P3t3HyPHfd95/lPVDzNkD8nhkzjTQ3FEPdqmGZEUSdtyFDKWNrJzcQKTFGJJh2VAZX1xdgMhuDMOwZ4gc6Fd3x6wOWj3Nl4ksbg501Ismk42OJ+d5fokJYqs2KJoR1o6lk1TtKme4aNmpruru+vx/mj2aDTqh+rq5+H7BRRUU1W/+v3qWzUU8PvO7/czJMM0FPiBnLyj/MW8spmsrMuW7Jwtt+jKd30FfiAF16a8MiUjVi6TeSXTangi6UTnb792KPdzZ2wl2VHZuqGygHUrC3yHiWm3v4dGC7n36/cp9eYbbbbOdnw3AAAAANCvGMkBYKAsXKuhkmyoNh1Rvesq/62VxKi1XkS9c6ju8ccfVyaTqRqnZqeXWrwGRzv2FUi+48vJOSpcLSg+HJdjOTIMQ27RVWmuJCfvyC268hyvnPDwgnLSo01qdTg2Gi3QSkdltbK16uulfk1w1FqYutMLVtdKBDTz7ppJcPRD3CvP168GJcFR7Vg//s4DAAAAQBQkOQAMnMWLVLd6XbUyzZ5Dbel0OvLxhft79+5t6356Z/neds5WYaagoUtDMgxDdtaWESsvNu7kHZXmSrLzthzr3ckOwzSU3lX9GdqhUSdkvY7fKB2v/dbp2U8d7bX0c9uqaTam7f7GlppBSHAAAAAAwPWAJAcALLIwIRI1sfH2mbf1vf/4PZ197qyunrkqSVpz6xpt/uXN2v17uzV602hb2oroxneMy7EcFWeKsi5big/F5bu+kiNJmXFTgR/ILbqy87ZKsyXZuWuJDsuVWyxvY9taW48D1Q1yR26/tr1f2zWoSHAAAAAAQP8gyQEAi4SZkqreVFUnPndCr37pVSmQhkeHtfqm1fI9X9YFSyf/+KROPXVKOz+7U/f+m3s79gwIZ8sDW3TmxJlyYiNWXng8OZKUmTClQPIcT47lyM6VEx2VUR3F2aK2Pri1J23uxTRDYUeVtKPeKFPxdKttrYjaznZM19SO6Y2i3qtV9Z691ZhG/WYGIcHR79N8AQAAAEA7sfA4ADSpXoLjy/d9Wae+dEpj28Z068dv1c333qyJ3RMa2z6msbvGNHbnmEY2jOjv/8Pf6+lPPN3llmOxA88ekGM5yk5llZvOKZvJKvtWVtlM+ef8hbysS5YKVwoqXC2oNFtScbYo+dK+r+zraNva3UHZ6H5h61t8XTvb2WrncSfbFua+YdsftZ3VruvHDvlmNYqn1L2Yhv096ecER63y7bwnAAAAAPQTRnIAGDiVJEOjdTbCXletTJTzR+8/qul/mNbk3kmtvW2tRsZHFB+Oy7M9lWZLsq5YslZYig3HZMQNnXvxnJ7+taelXaGbhw449OIhHb3/qLLns3IsR4lUQvFkXDKlwAvkOZ5cy50fwREEgR556ZG21d+oU7WdnZBhOn5r1bfwL8Or3afVdi68Z6O/3m9X28J0rtcqHyWBErWdjcrV0kpMo4oa06gJqU7FtNE7bzae7YhLu38vAAAAAGCpYCQHgIFSSTBUEheLp5YKe13l+ML9ynWVY4sTGovLLCz3w+M/1PnvnNfE7gnd8MEblN6Z1sYPbdTYtjGtvX2tVk2u0sqJlUptSCm1PqXla5drZHxEbz7/pvSjtoQGEaV3pfXwtx5W7nJOM2/OlEdzZLLKTeXmR3PkL+WVncrKyTk6eOKgxnZ0fi2Oc4+2t/O50b3C1Fevw7jXutm2enV1Koat1Nnv6rW/VzFdKpbydwMAAAAAFYzkADBwao2kWHw87HVR77/Qc489p9WbV2vVplVac8sarf/geq29fa0KVwrKZrIK/ECe7ckpOOU1HvJ2eZ2HVEnOtx3p9rpNQodN7J7QY6XHdPzB43r9q68rPhxX4F9LlJmG3KKrrQ9uDT1FVZjOw252vrezzk51jPaiba3U2Yuy3Xy+qPfs1rP1ot5BeOedugcAAAAA9DOSHADQotxUTld/elWT90xq2eplGhodUjKVlCSZcVOxoZjiw/H3bsviSqQS0s8lIx9+Si10zv5n9mv/M/s1fWpamVcyksojPca2dX7kBgAAAAAAAJpHkgMAWpQ5mZlPZhgxQ77rqzhTlAzJd325RVe+60vBtamuTENmzJQZL2+GaSjI1B9dgu4a2z6mse0kNgAAAAAAAPoda3IAGAgvvPCCTp48+a6t4gtf+ML8/uHDh7u+P/PTGZkxszwlVam8yHhuOqdsJivroqXSTElO3pFbdOU5ngIvKE+FdC3pETNi0tXmYwL0s8knw28AAAAAAABRMZIDwEB4/vnnFQSBPv/5z7/n3B/8wR/M7z/++ONd3x+9ebQ8YqPgqjRXknXZkhkzZedsmTFTnu3JzpXX4HCsd5IdvuvLd315vietCR0KAAAAAAAAANeQ5ACAFqXvSsv3fJXmSipcLSixPKHAD5TMJmXGTQVeIKfgyM7a5S1vy7XccrLD9hT4gYI001VhaWGxYwAAAAAA0A0kOQCgRSPjI1p7y1oVLheUX5WXGTflOZ6SI0nFErHyNFa2Jztvy567tl0b1eEUHGmdpFSvnwIAAAAAAAAYPCQ5AKANPvaFj+n4Q8eVWJGQYRhyS+58kkOSfMeXU3Rk52yV5koqzhVl52y5BVf6VI8bDwAAAAAAAAwokhwA0AZ3/PodumnPTTr3N+cU+OXpqZKppGLJmGRIgVcezeFY1xIdsyVZVyzd8k9u0Y9u+5HEbFUAAAAAAABA08xeNwAAloqHvvGQNv3iJs3+bFa5TE7ZTFbZTFa5qZxyF3LKX8grfykv67Kl/OW8brn3Fn36v3y6180GAAAAAAAABhYjOQCgjR7+5sP69r/8tl75o1eUu5BTbCim+FD5n1q36MoreYoti+mjn/uo9h7e29vGAgAAAAAAAAOOJAcAtNm9//pe7fzMTn3vj76ns98+qys/viJJWnP7Gt18783a/S92a+XGlT1uJQAAAAAAADD4SHIAQAesmlyl+/7tfb1uBgAAAAAAALCksSYHAAAAAAAAAAAYSCQ5AAAAAAAAAADAQCLJAQAAAAAAAAAABhJJDgAAAAAAAAAAMJBIcgAAAAAAAAAAgIFEkgMAAAAAAAAAAAwkkhwAAAAAAAAAAGAgkeQAAAAAAAAAAAADiSQHAAAAAAAAAAAYSCQ5AAAAAAAAAADAQCLJAQAAAAAAAAAABlK81w0AALzDMIz5/SAIIl0X5dzC42HqBwAAAAAAAPoBIzkAoE9UEg2V5EK1xEOj6wzDUBAE81vYcwuPk9wAAAAAAADAoGAkBwD0kUqCYXESIux17UhQVJIhjUydnFLmZEaSlN6Z1viO8arXTT4ZrR3nHm1c9tyjzd1z4f2aKdtsuWrtbratzdy7mftHbdvict2ur5N1thrTWvdqVL7ZmLb6+9Ctd9HubzTq704n3wUAAAAAoH+Q5AAANOXYgWM6ffy0EssTkiHpWj7EsRxteWCLDjx7oGttmXyytY7vdperd23lXCudp63ev1b5enHsRNwavbco7YxaZzvfWdhYRY1pK9od007Es51x6ed3AQAAAABoL5IcALBELR6R0Wh0SLUyC51/6byO7Dmi1LqUNt2zScmRpAzTkO/4snO2ijNFnTlxRk8MPaFDLx5SeldaUuMOzTAdyPU6U5tJdHRSo78ab1d7G8UzTOfzwvNh21U512yn8OJ7drqd1epcfN8w1zdbZxTtimkjUWMatVyr8Ywal1b0ok4AAAAAQHuwJgcALEG1khVR1904/9J5Pf3JpzW+Y1yb79usid0TSu9Ma2zbmNa/f73W3LZGo5tHtfb2tUptSOno/Uf11nffatfjVBVluqmoncPNlKt1bTs6ycN0DDd7jzAJg3bGrVPtbPSuoryXTn1jUWLaDs3GNEq5VuLZrrgMwrsAAAAAALQPSQ4A6COVkRZhRlzUui7Mmhph192oOLLniNbcukbjO8aV3pnWxg9v1Phd41r/gfVafetqjU6OauXESo2MjWjF+AolliX01D1Phb5/N0TtyFwKHaBhEwDd/iv2xfX1azvD6sdvJWpMeRcAAAAAgEHBdFUA0Ccq00lVEhcLkxALkxKNrlv434XnGy1QXivxcezAMaXWp7T29rVa9751GtsxptHJUXm2p/zFvOLDcRmGId/15dme3KIrx3JUnCvq65/+uvb9+b7IMWmHbqzFgd5qx3on7ai/V/XRof8Ofm8BAAAA4PrDSA4A6CO1ppOq9nOt6xZvjcrUqqPi9PHTWn3raqU2pLRiYoVGJ0e1atMqLV+3XMOjwxpaOaTkSPKdLZVUIpXQ8Ophvfbsa1HCEEqYzsxeTFNVb6HmKG0Ji87dd0w++e6tlfuEvaaXiYZWn7NbOt3GfngXAAAAAIDuYyQHAKCmqZNTSixPKDmSVGJ5QmbClGd7Ks4U5RZdBf61USUxQ2bclJkwZSZNxZPx8jYc1/SpaY1tH2upHVEWjQ57vp3lzj1av8O5G52v/dDBuzAOtRYe74ROJZg69Y1F0c14dkon49UP3z8AAAAAoLsYyQEAfeCOO+7Q1NTU/Fax8Nji43Nzc3rhhRfmj3Vi//lnnpcMyTANBX4gJ+8ofzGvbCYr67IlO2fLLbryXV+BH0jBtWmxTMmIlctkXsm0EJnorqdpqvr5L9jbNaqiGZXFpFtZVDpMTLv9rTRayL2fv11GMgEAAAAAOoWRHADQY48//rgymUzV6aIaTS21eDqqdu+XD0i+48vJOSpcLSg+HJdjOTIMQ27RVWmuJCfvyC268hyvnPDwgnLSo01qdYw2Gi3QjWmqwpav1dZW9WuCo9b0XZ1esLpWIqCZ+DeT4OiHuFeer191K8HRD+8CAAAAANB9JDkAoA+k0+lIx/fu3Tt/rCP7D+3V2S+elZ2zVZgpaOjSkAzDkJ21ZcTKi407eUeluZLsvC3HeneywzANpXdVf4Z2aNR5Xa/jt1FCotlyjTpam+1oD2MQOnf7uW3VNBvTqN/Y9aKbMeBdAAAAAMD1iSQHAKCm8R3jcixHxZmirMuW4kNx+a6v5EhSZtxU4Adyi67svK3SbEl27lqiw3LlFsvb2LbW1uNAdYPcaduvbe/Xdg0q4gkAAAAA6AaSHACAurY8sEVnTpwpJzZi5YXHkyNJmQlTCiTP8eRYjuxcOdFRGdVRnC1q64Nbe9LmqNMMDcr0RM22JeyIl14/W9R2tmO6pigxbde9WlXv2VuNadRvppsx6Kd3AQAAAADoPhYeBwDUdeDZA3IsR9mprHLTOWUzWWXfyiqbKf+cv5CXdclS4UpBhasFlWZLKs4WJV/a95V9HW1bP65DUKtN7Wpru9YNqfVzuzSKQ6P2R21ntev6qUM+qjDfVbdi2qjcIMQTAAAAALB0MJIDANDQoRcP6ej9R5U9n5VjOUqkEoon45IpBV4gz/HkWu78CI4gCPTIS4+0rf5Gnar90Jm6cDRBo7+sj2rhfZuto1H7GiUAGh1vdm2TWqK2M2z8F2slplFFjWnUhFSnYtronTcbz1a+tah6UScAAAAAoL0YyQEAaCi9K62Hv/WwcpdzmnlzpjyaI5NVbio3P5ojfymv7FRWTs7RwRMHNbaj82txnHu0vzof67WnH9par23drKtRfVHb2c+xb1W99vcqpgAAAAAA9ANGcgAAQpnYPaHHSo/p+IPH9fpXX1d8OK7ADyRJhmnILbra+uDW0FNUhekk7WbnezvLdaoDuB33bfYerdTZi7LdfL6o9+zWs/Wi3l59L1HvSbIGAAAAAAYfSQ4AQFP2P7Nf+5/Zr+lT08q8kpFUHukxtq3zIzcAAAAAAACAhUhyAAAiGds+prHtJDYAAAAAAADQOyQ5AADokWYWkmZaHQAAAAAAgPdi4XEAAAAAAAAAADCQGMkBAECPMDoDAAAAAACgNSQ5gCXKMIz5/SAImr4uTPlG1xiGUbduAAAAAAAAAGgF01UBS1Al+VBJMCxMRoS9LgiChsmRyjW1EhwAAAAAAAAA0EmM5ACWqEriIQiCugmHsNct1GiERuV8o/v5jq/v/+fv6yff+oku/MMFSdKGOzfo1o/fqm2/tU1mnDwsAAAAAAAAgNpIcgCIpN40V2GmqDr1pVP61u9/S5KUWJZQciSpwA/05nNv6id//RP99e//tX71//pV3XnwzvY3HgAAAAAAAMCSQJIDQCTVEhthExzf+Ow39NrTr2nD1g1KbUgpOZKUIUNuyVVprqTC2wUVrhT0jX/+DU29OqWPP/nxTj4KAAAAAAAAgAFFkgNAWy2eompx4uMvD/6lfvRXP9KmezZp7a1rNTI+oviyuHzHV2muJOuKpaFLQ0oMJ2TGTb36pVflWI60sdtPAgAAAAAAAKDfkeQAlqiw62KEvS6MxaM4Fic4zn/nvH54/Ie68Rdv1IatG7R+y3qt3LhShmmUExyXLMWXxefX4vA9X57j6bWvvCb9j5LSLTcRAAAAAAAAwBJCkgNYgipJi0riotaaGY2uW7xfa5HyMFNUSdI3f++bWrVplUYnR7Xm1jXa8AsbtObWNSrOFpWbykmSPMeTW3LlWI7svC07b6s0W5Lz/zrSbzcdCgAAAAAAAABLmNnrBgDojCAI5rfFx8Net3gLU65WXYUrBU2/Nq2R8REtW7usvK1epsTyhBLDCcWXxZVYlihvw4l39q8tSq5pScWIwQAAAAAAAACwJDGSA0BXZE5mFIvHFF8WVywRU+AHKs4WZcQM+Y4vr+Qp8APJkAzTkBEzZMbN8pYwZcQMBZlwI0YAAAAAAAAAXB8YyQEsIdu2bdMnPvEJvfbaa/NbxWuvvaaXX355/ucnn3yyq/uX//GyYvGYFEhuyVVprqT8hbyymazyl/IqzZZkW7bckivf9RX474wSMQxDMSMm41Lr64YAAAAAAAAAWDoYyQEsIb/xG78h27arnrvjjjvkOM78zwcPHuzq/tT/NyXP9eQWXdlZW9ZlS2bclJ23ZZiGvJInO2fLztlyLEdeyZPv+PK98ub5nrQqdCgAAAAAAAAAXAdIcgBLTDKZrHl84bnR0dGu7ht3GQr8QKW5kgpXC0qkElIg2VlbZtyU7/nlxcazdnnL23IKjryiJ9/2y6M60qHDAAAAAAAAAOA6QJIDQFesvHGlVoytUPFKUdYqS7FETL7jKzmSlJkwFfiBvJInx3JUmi2pNFeaH9VhF2xphaSVvX4KAAAAAAAAAP2EJAeArjBMQ3f/r3frxOdOKDGSkEzJtV0lU+UkhyT5ji+34MrO2+9KdLiWK/0TKRALjwMAAAAAAAB4B0kOAF2z83d26tSXTunKj68oCAK5xXKSI5aISYbKa29cG81h58qJDuuypQ13btD57edFjgMAAAAAAADAQmavGwDg+vLPvvvPtHLjSr199m1l38qWt6msctM55afzyl/My7psybpiKXcxp9W3rdYj33mk180GAAAAAAAA0IcYyQGguwzpd1//XR174Jh++t9+qvzFvBLLEooNxaRAcoqO3IIrc8jU+37jfdr/zP5etxgAAAAAAABAnyLJAaAnHjj2gM79zTl95999R2999y1lM1lJUmpDSps+ukl3f+5ubfrFTT1uZfcZhjG/HwS15+eqd12tcwuPN1MOAAAAAAAA6FckOQD0zOQvTWrylyYllRcdlzS/CPn1qJJkCIJAhmHIMIyqyYZ61y0us/jnWsmLRuUAAAAAAACAfkSSA0BfuJ6TGwtVEguVBEaz10VNTEQpN3VySpmTGUlSemda4zvGq143+WSkJunco43Lnnu09rlaZeuVqVc+bLladTdTvtl7N3P/qG2LGo921dfJOluNaa17NSrfbExb+X2oVb4T76Lbv3th/41pps3t+n0FAAAAAHQeSQ4AWGKiTGXVjGMHjun08dNKLE9IhqRrt3EsR1se2KIDzx6IdN8oJp+s3hlZr9Ozcq5WJ2bUpEyYelvtOG3lueqVr9e2qPFoJRZR2hm1zlZjGvZeUa5rp3bHtN9+96LqRZ0AAAAAgPYiyQEAS0ytaaeqJTxqTYdV7fj5l87ryJ4jSq1LadM9m5QcScowDfmOLztnqzhT1JkTZ/TE0BM69OIhpXelJTXu0AzTgVyvM7VeJ23UcgvSXzbsAAAgAElEQVTLRukEXXzfsHVGuXfYOmqNMOh0PJqNRavtrFbn4vuGub7ZOqNoV0wbiRrTqOVajWezcQmTpAn7zZD0AAAAAIDBw/wwAIB59RIcT3/yaY3vGNfm+zZrYveE0jvTGts2pvXvX681t63R6OZRrb19rVIbUjp6/1G99d23OtrWRp2kzSQ+wpatp15nars6yMN0DDd7j07Eox2xaLadjTqzo3wPUaYoC5u068V0SM3GNEq5VuLZi7j06l0AAAAAANqHJAcA9JHKdFL11uOod12jco3uWWsKqyN7jmjNrWs0vmNc6Z1pbfzwRo3fNa71H1iv1beu1ujkqFZOrNTI2IhWjK9QYllCT93zVOS2LGW9nJKnUUd/t9u2uL5+bWdY/dhZHjWmg/4upP5uGwAAAACgfZiuCgD6RGUR8Uqiot60U7Wuq7cQ+eIESLVz1coeO3BMqfUprb19rda9b53GdoxpdHJUnu0pfzGv+HBchmHId315tie36MqxHBXnivr6p7+ufX++r7XAAA00u3ZGp+rvVX39mFzpJ8QHAAAAAJY2khwA0EdqjaRYfLzeouFh7xH23Onjp7Xpnk1KbUhpxcQKjU6OatWmVSrOFOXZnhzLkZ21lRxJlrdUUolUQsOrh/Xas691LMnR6oLYS63jk79af0e7kgBhYtoP31M/tCEMvlEAAAAAQCeQ5AAA1DR1ckqJ5QklR5JKLE/ITJjybE/FmaLcoqvAvzaqJGbIjJsyE6bMpKl4Ml7ehuOaPjWtse1jLbUjyqLR9cp2ahHpySerL6rczc7dfujo7lUsat271SRAlIXOO6WX31a7dCNeg5L4AQAAAAC0jiQHAPSBqampd/08Pj7e1PGzZ8/q7rvvliS98MIL2rNnT1v2R340IhmSYRoK/EBO3lH+Yl6e7SnwA9k5W27Rle/6CvxACq5NeWVKRqxcJvNKpuUkR7tV63xv9/27qZ87dHvRCd+ORECYmHb72Wot5F5pR6e/61b08zcKAAAAABhsJDkAoMcOHz6s2267TXv37n3PuSjTV7V9P5B8x5eTc1S4WlB8OC7HcmQYhtyiq9JcSU7ekVt05TleOeHhBeWkR5vU6hitNVqgUdkw5aKotRhzJxdp7tfO417EYuH9Fx9r5p03k+Doh7gvTHT0o36KFQAAAABg6SHJAQB94I033tBDDz30nuPpdLrq9YuPL/x5YbKk1f2pleURI3bOVmGmoKFLQzIMQ3bWlhErLzbu5B2V5kqy87Yc693JDsM0lN5V/RnaodnO61bLNXP/bhiEzuN+bls1zca0XnJhEN5Pp/UiBsQdAAAAAK4vJDkAADWN7xiXYzkqzhRlXbYUH4rLd30lR5Iy46YCP5BbdGXnbZVmS7Jz1xIdliu3WN7GtvXXVFW90u6O10HuyO3XtvdruwYV8QQAAAAAdIPZ6wYAAPrblge2KJvJyrpkKTedUzaTLW9TWeWmc8pfysu6bKnwdkGl2dL8qI7ibFFbH9za6+YvSc12HjeaIqpfOqOjtrMd7Y4S01rb4ms6rd5oklZjGvWb6dU31c/TdgEAAAAAOoMkBwCgrgPPHpBjOfNJjWwmq+xb5URHbjqn/IW8rEuWClcKKlwtJzqKs0XJl/Z9ZV9H29aoA7bZcq3qRhKh1XstbmO/xiJqO6td168d8s0I8613K6Zhf796Gc9+fpcAAAAAgPZiuioAQEOHXjyko/cfVfZ8Vo7lKJFKKJ6MS6YUeIE8x5NrufMjOIIg0CMvPdK2+ht1qtZaXLyRZsrVWkA7Sr1RLbx3mL/eX3ysUqZa2WafJ0w8osQiajsblaullZhGFTWmUb+tTsW00TtvNp6t/u5F0Ys6AQAAAADtxUgOAEBD6V1pPfyth5W7nNPMmzPzU1blpnLzoznyl/LKTmXl5BwdPHFQYzs6vxZHremAGnVIdmoaoXqdxf3QSVqvfd2sK8z7aeZ4O+rsd/Xa36uY9pt+GEECAAAAAOg+RnIAAEKZ2D2hx0qP6fiDx/X6V19XfDiuwA8kSYZpyC262vrg1tBTVIXpiGylszJq2VY7SDvZwdqOezd7j168g1bKdvP5ot5z0L7NZsr16nuJWp6ECAAAAAAMPpIcAICm7H9mv/Y/s1/Tp6aVeSUjqTzSY2xb50duAAAAAAAAAAuR5AAARDK2fUxj20lsAAAAAAAAoHdIcgAA0CPNLCTNtDoAAAAAAADvxcLjAAAAAAAAAABgIDGSAwCAHmF0BgAAAAAAQGsYyQEAAAAAAAAAAAYSSQ4AAAAAAAAAADCQSHIAAAAAAAAAAICBRJIDAAAAAAAAAAAMJJIcAAAAAAAAAABgIJHkAAAAAAAAAAAAA4kkBwAAAAAAAAAAGEgkOQAAAAAAAAAAwEAiyQEAAAAAAAAAAAYSSQ4AAAAAAAAAADCQSHIAAAAAAAAAAICBRJIDAAAAAAAAAAAMJJIcAAAAAAAAAABgIMV73QAAaJZhGPP7QRA0fV2j8vXOh60bAAAAAAAAQOcxkgPAQKkkGSoJhoVJh7DXBUFQM0FhGMb8+SAI3lWu3jkAAAAAAAAA3cdIDgADp5KgaJRoCHtdtTLNnnvvxdKbL7yps98+q/Mvn5ckbbx7ozb/8mbdtOcmifwIAAAAAAAA0DKSHACwSKvTVf38736urz34NRVnijJMQ8nlSQV+oPMvn9fLf/iyhtcM6zef/U2lP5TuzAMAAAAAAAAA1wmSHACwyOL1Oxb+XO+cJP3d//F3+pt/9TcavXlUG7ZuUHIkKcMw5JZc2VlbhZmC8hfyOvKxI7rv39ynDz36oc4/EAAAAAAAALBEkeQAgDZ54V+9oJf/8GVtvHuj1ty6RiNjI0osS8h3fZWyJRWuFjR0eUiJZQnFl8X13P/2nBzL6XWzu6bVBeMbnWfBeAAAAAAAgOsPSQ4AA6cygqLROhthr6tWptlzM2dn9Ldf+FttunuTNvzCBq3/wHqtnFgpI2bIztqyrlhKppKKJWKSpMAP5Lu+nj/8vPTPJa0I3cSBtHAheMMwasay0XX1ytW6Luw9AAAAAAAAMHhIcgAYKAs7vys/VyzsvG503eL9WouUL56qqta5v/inf6HRG0c1unlUa29fq7E7x7T65tWyc7ZyF3Iy4oYCL5Bne3KLrtyCKyfvyJ6xZX3dkg62Hpt+1+qC8VGTE1HKTJ2cUuZkRpKU3pnW+I7xqtdNPtn0rSVJ5x5tXPbco9WPh62zVvla96h3fbNqtTFsHVHbt7hct+vrVJ2txrPe/Rrdo9mYRv2u65XvxLto9zfazLvoxfcGAAAAAEsZSQ4AA6dWp/Xi42Gva+Z8tXN2ztb5l89r0z2btHzdci1fv1ypG1IaXj0sGVIil1ByeVKJ5Yl3tmUJxZfHlViZkH4mGW740SbXs05PO3XswDGdPn5aieUJyZB0rQrHcrTlgS068OyBttdZy+STnenArNW526766nVyV85F6Xyu176oyaZGbW13O6PU2Wo8m6k/ynXt1O6Y1irXSkxbiUvU762V7xQAAAAArgckOQCgRVMnpxQbipUTF0NxGTJUypWUv5CXZ3vyHb/cIW9IhmnIiBkyE6ZiiZhiiZiMmCH/Lb/XjzEQGo3caaTWaJDzL53XkT1HlFqX0qZ7NpUXjDcN+Y4vO2erOFPUmRNn9MTQEzr04iGld6UlNe6UDNP5WK8TtloHZpjO3jCdpQuvqVdfFI3qD9P5HKV9lXPNdkTXSix0qp3V6lx830bXNltfVO2KaSNRYxq1XKsxjRqXWnWHuU+z3ykAAAAAXC/MXjcAAMK46aab9LGPfUxvvPHG/Fbx7LPPzu//2Z/9Wdf3L/zDBZmmKRmS53gqZcsJjmwmq/zlvEqzJTmWU054eL4CP3hX0sM0TRkXGMnRqiAI5rdq6iU4nv7k0xrfMa7N923WxO4JpXemNbZtTOvfv15rblszPw1ZakNKR+8/qre++1ZHn6XTHZbNJE5avXeUeppt37lHo3eqN9MB3ui6sFMx1bqu2ba085mr3bsXHedRv81myrUS06hxifLuG5UjsQEAAAAAZSQ5AAyEgwcPavv27RofH5/fKj7ykY/M7+/Zs6fr+8vXLZfv+XKLruysrcKVgnLTuXKSYzov64ql0mxJds6WW3DfGd3hBgq8ax3zy5f+QtiVkRZhFowPc1298ouTGfXW8ziy54jW3LpG4zvGld6Z1sYPb9T4XeNa/4H1Wn3rao1OjmrlxEqNjI1oxfgKJZYl9NQ9T0VqWyc1+kvwsJ2svZimaGG9/da+xfX1azvD6seO8agxHfR30U7XwzMCAAAAQC1MVwVgYKxYsaLq8RtvvHF+/6abbur6fnpnWkEQlBMcVwtKjiTLP6+wy1NRub7cgqvSXEmlbElO3ikvPl5y5TmefN+XMbG0R3K0Y8H4egu/11uro1rSpHLNsQPHlFqf0trb12rd+9ZpbMeYRidH5dme8hfzig/HZRjld1hZNN6xHBXnivr6p7+ufX++r7XAdEA/dmIj2voZnai/V/Vdz99lr989AAAAACx1JDkAoEWjm0eVHEmqeLUo67KlWDIm3/VVypYUi8cU+IHckisn78wnOuy8LafgyCk40pCk0V4/Ree1umB8M/cIe+708dPadM8mpTaktGJihUYnR7Vq0yoVZ4rybE+O5cjO2kqOJMtbKqlEKqHh1cN67dnXOpbkWKp/lb1UnyuKdiQBml1EvJed7P3QhjC68Y2SAAIAAACA9iLJAQAtMuOmdn12l77zh99RYiQhwzTk2Z4SqYRiyZgUlNfqcAuu7Jyt0mxJpbny9FVOzpE+IgXG0p+uqt9MnZxSYnlCyZGkEssTMhOmPNtTcaYot+gq8K+NKokZMuOmzIQpM2kqnoyXt+G4pk9Na2z7WEvtqNepGrbzc1A6kCv6oZ3nHi3HrdqizZ3s6K5171beYdQF7julm/HslE7EK8q779V3CgAAAACDhCQHALTB3sN79YP/+weaOz8nBZJbdOeTHJXpjtySOz8yoDRbUuFqQSsmVujqL12VlnCOY2pq6l0/V9ZT6cbxZcuW6Qc/+MH8+ikvvPDC/P7zzzw/v/h74Ady8o7yF/PybE+BH5TXUCm68t3yYvEKrk15ZUpGrFwm80qm5STH9aKfEzG96CxutcM6bDy7/Wz1Ouor7enHb0Dq3jca9d2T1AAAAACA6lh4HADa5LOvf1axZEwzb84om8m+s01llbuQU/5iXtYlq7ww+cWchlYO6Xd+8Du9bnZHHT58WM8991x5cfVrW8XCY506vvC/i/fLByTf8eXkHBWuLlgw/kJehSsFlebeWUPFc7xywsMLykmPNjn3aPVNWjqdmv2a4FgY6zDH21lvrWNh3nmzCY5+iHs/tKGeXiU4Fh6r9e579Z0CAAAAwKBgJAcAtEkyldTv/+z39Scf+hNd/fFVGaZRHs0RjykIAvm2L9uypUC6YcsN+u2Xf1ta2uuNS5LeeOMNPfTQQ+85nk6nq17f7uN79+6dP/au/Yf26uwXz8rO2SrMFDR0aUiGYcjOvrNgfGUdFTtvy7HenewwTEPpXdXrbod609Qs1E8d2dX0e/uk/m7bYlHiWS9xMgjvp9MGJQb93j4AAAAA6BWSHADQRkbM0Gde+Yx+8J9/oJf+3Uu68sYVmYnyoDnf8bX2jrW6+3+5W3f+0zt73FKM7xiXYzkqzpQXjI8PxeW7vpIjSZlxs7xgfNGVnS9PL2bnriU6LFdusbyNbWOqqnoGpfO4mn5sez+2adANekwHvf0AAAAA0A4kOQCgA+78rTt152+VExlXf3xVkrTmtjW9bBKq2PLAFp05caac2IiVFx5PjiTLialrC8Y7lvPuBePztoqzRW19cGuvm9/UdFaNRoa0u7O02ft1u31RRW3nwnUpoojy/P00Aqjes7ca06jfTDenqFoqU88BAAAAQD9iTQ4A6LA1t60hwdGnDjx7QI7llNdNubYeR/at8loquemc8hfeWUelcLWg0mxJxdmi5Ev7vrKvo21rNoHRyr3b3QHbaudxp9vX6L5RF/VuZQHpenX2S4KnkUbxlLoX00blehXTZt99rTJhygEAAADA9YKRHACA69qhFw/p6P1HlT2flWM5SqQSiifjkikFXiDP8eRa7vwIjiAI9MhLj7St/kadse3swFz4F+XV6m1HXQvv2+iv96sdi9K+MJ3rtcpHSaBEbWejctW0Es9WRI1p1IRUp2La6J03G9OocYny7sPUCwAAAABgJAcA4DqX3pXWw996WLnLOc28OVMezZHJKjeVmx/Nkb+UV3YqKyfn6OCJgxrb0fm1OM492pnphup1FveDbravXl2N6ovazlbq7Hf12t+rmPaTqO9+KX8zAAAAANAOjOQAAFz3JnZP6LHSYzr+4HG9/tXXFR+OK/ADSZJhGnKLrrY+uDX0FFVhOh5b7ZxspXwnO0bbce92JW76tWy71tVoRdTEQav37VT5bsV0EJ4PAAAAAK43JDkAALhm/zP7tf+Z/Zo+Na3MKxlJ5ZEeY9s6P3IDAAAAAAAAzSPJAQDAImPbxzS2ncQGAAAAAABAvyPJAQAAampmwWOm1AEAAAAAAN3GwuMAAAAAAAAAAGAgMZIDAADUxOgMAAAAAADQzxjJAQAAAAAAAAAABhJJDgAAAAAAAAAAMJBIcgAAAAAAAAAAgIFEkgMAAAAAAAAAAAwkkhwAAAAAAAAAAGAgkeQAAAAAAAAAAAADiSQHAAAAAAAAAAAYSPFeNwAA8A7DMOb3gyCIdF2UcwuPh6kfAAAAAAAA6AeM5ACAPlFJNFSSC9USD42uMwxDQRDMb4vP1Sq3sAzJDQAAAAAAAAwKkhwA0EcqCYZGiYZa10Utt1AlUQIAAAAAAAD0O6arAgA05e0zb+t7//F7OvvcWV09c1WStObWNdr8y5u1+/d2a/Sm0R63EAAAAAAAANcLkhwAsERFGZHRqMyJz53Qq196VQqk4dFhrb5ptXzPl3XB0sk/PqlTT53Szs/u1L3/5t5Wmw8AAAAAAAA0RJIDAJagTkw59eX7vqypV6c0tm1MIxtGlEwlJUlO0VFprqTi1aKsy5b+/j/8vS6cuqCHvvlQW+sHAAAAAAAAFiPJAQB9pJKcqLXoeJjr6iU4wt5/saP3H9X0P0xrcu+k1t62ViPjI4oPx+XZnkqzJVlXLFkrLMWGYzLihs69eE5P/9rT0q6mqgEAAAAAAACaQpIDAPpEJflQSUAsTFQsTFw0um7hfxeer1ducR0L/fD4D3X+O+e16Zc26YYP3qANWzdo5caVkiEVZ4rKX8wrPhyXYRoK/EC+48uzPb35/JvSSkm3txgYAAAAAAAAoAaSHADQR2qNwFh8POx1zZyvde65x57T6s2rtWrTKq25ZY3Wf3C91t6+VoUrBWUzWQV+IM/25BQcOZYjO2/LztkqpUpyvu2Q5AAAAAAAAEDHmL1uAACgf+Wmcrr606tafsNyLVu9TEOjQ/NrcZhxU7GhmOLD8fduy+JKpBLSVcnINzc1FgAAAAAAABAWIzkAADVlTmbmkxlGzJDv+irOFCVD8l1fbtGV7/pSUJ7uyjANmTFTZry8GaahINPeBdABAAAAAACACkZyAECP7d27V5/85Cd18uTJ+a1i4bG//du/nT9++PDhruzP/HRGZswsT0lVKi8ynpvOKZvJyrpoqTRTkpN35BZdeY6nwAsU+MF80iNmxKSrkUMzMCprnYRZML7RdbUWk69VLmzdAAAAAAAAS5ERNJrAvUWHDx9WEAT6/Oc/38lq0GZL+b0t5WcD2u2N/+cNfe03v6aJ3RNa9751Gt08qpENI0quSMqMmfJsT3bOVnGmKOuKJeuSpfylvPIX8spN5zTz8xkFDwR6/CuP9/pROmbhQu61FnUPe12jRecX/1zvHAAAAAAAwPWA6aoAADWl70rL93yV5koqXC0osTyhwA+UzCZlxk0FXiCn4MjO2uUtb8u13PLIDttT4AcK0ku/072SWFiYwGj2ukqCotMjMqZOTilzMiNJSu9Ma3zHeNXrJp+Mdv9zjzYue+7R6sfD1lmrfLV71Ls2ilptDFtPtfJhykZ9rnbV16k6W41nvfs1ukezMY36Xdcr34l30e5vNEpdzdbdyvcGAAAAACQ5AAA1jYyPaO0ta1W4XFB+VV5m3JTneEqOJBVLxMrTWNme7Lwte+7alrPlWI6cgiOtk5Tq9VP0v26MwDh24JhOHz+txPKEZEi6Vp1jOdrywBYdePZAR+tfaPLJ7iUfulVH5VyUDuF68Yj6XI3a2u52Rqmz1Xg2U3+U69qp3TGtVa6VmPYiLvXq7cS/EwAAAACWJpIcAIC6PvaFj+n4Q8eVWJGQYRhyS+58kkOSfMeXU3Rk52yV5koqzhVl52y5BVf6VI8bPwAaJTjqje4IM/Lj/EvndWTPEaXWpbTpnk1KjiRlmIZ8x5+fauzMiTN6YugJHXrxkNK70pIad4SG6Xys1wlbrQMzTGdvo3or5zvVYdsoLmE6nxeeb1RucZlmn6tWYqFT7axW5+L7Nrq22fqialdMG4ka06jlWo1pM3Fp9Xe2Hd8bAAAAALDwOACgrjt+/Q7dtOcmzZ6bVXYqq2wmq+xb1/47lVXuQk7WJUuFKwUV3y6qNFOSdcXSzffdLN3W69YPhsWLhy9OXARBML8tVu/c+ZfO6+lPPq3xHePafN9mTeyeUHpnWmPbxrT+/eu15rY1Gt08qrW3r1VqQ0pH7z+qt777Vmce8ppOdlh3ujM0TMdws/cIkzCI2qneTAd4o+vCTjdU67pm29LOZ6527150nDcb0yjlWolpr+JSqbvezwAAAABQD0kOAEBDD33jIW36xU2a/dmscplcOcGRySo3lVPuQk75C3nlL+VlXbaUv5zXLffeok//l0/3utldUys5Eea6hUmKhWt21CrfzLkje45oza1rNL5jXOmdaW388EaN3zWu9R9Yr9W3rtbo5KhWTqzUyNiIVoyvUGJZQk/d81S4h+6iXk2j0y5hEwDdfs7F9fVrO8Pqx47xqDEd9HcRpl2D/owAAAAA+gfTVQEAQnn4mw/r2//y23rlj15R7kJOsaGY4kPl/424RVdeyVNsWUwf/dxHtffw3t42tosqU0ZVEhcLEw0LEw/1rqtncUIk7LljB44ptT6ltbev1br3rdPYjjGNTo7Ksz3lL+YVH47LMAz5ri/P9uQWXTmWo+JcUV//9Ne178/3NRGF7ujHTmxEWz+jE/X3qj6+y9qIDQAAAIBuIMkBAAjt3n99r3Z+Zqe+90ff09lvn9WVH1+RJK25fY1uvvdm7f4Xu7Vy48oet7L7aiUsFh8Pk9hopky9c6ePn9amezYptSGlFRMrNDo5qlWbVqk4U5Rne3IsR3bWVnIkWd5SSSVSCQ2vHtZrz77WsSTHUv2r7KX6XFG0IwnQ7CLivexM74c2hME3CgAAAGCpIskBAGjKqslVuu/f3tfrZqCOqZNTSixPKDmSVGJ5QmbClGd7Ks4U5RZdBf61USUxQ2bclJkwZSZNxZPx8jYc1/SpaY1tH2upHfU6VcN2CA9KB3JFP7Tz3KPluFVbtLmTHd217t3KO4y6wH2ndDOendLpeA3a7ywAAACAwUeSAwCAiKampt718/j4eNuOv/DCC9qzZ48kNb3//DPPS4ZkmIYCP5CTd5S/mJdnewr8QHbOllt05bu+Aj+QgmtTX5mSESuXybySaTnJcb3o507dXnTCt5oICBvPbj9brcXTK+2ollTqF/38jQIAAABAq0hyAAAQweHDh3Xbbbdp79697zkXdvqqescXHmt2v3xA8h1fTs5R4WpB8eG4HMuRYRhyi65KcyU5eae8norjlRMeXlBOerRJrQ7VWqMMBlG/dh7XWrS504s510sEhHnnzSY4+iHuCxMd/aifYgUAAAAAnUCSAwCAiN544w099NBD7zmeTqerXt/M8YXJk6b3H9qrs188KztnqzBT0NClIRmGITtry4iVFxt38o5KcyXZeVuO9e5kh2EaSu+q3tZ2CNvp3e+ds/3ePqm/27ZYlHjWSy4MwvvptG7HgJgDAAAA6AWSHAAALDHjO8blWI6KM0VZly3Fh+LyXV/JkaTMuKnAD+QWXdl5W6XZkuzctUSH5cotlrexbUxVVc8gd+b2Y9v7sU2DjpgCAAAAuF6YvW4AAABovy0PbFE2k5V1yVJuOqdsJlveprLKTeeUv5SXddlS4e2CSrOl+VEdxdmitj64tdfNX1LT/zSaIqpfOqOjtrPVdkd5/nOP1t4WX9Np9b7VVmMa9ZvpxTfV7O/soPxeAAAAAOh/JDkAAFiCDjx7QI7lzCc1spmssm+VEx256ZzyF/KyLlkqXCmocLWc6CjOFiVf2veVfR1tWzOdof3Wwdlqx+viZ+9UMqfVjuOo7ax2Xb06B6Uju1E8pe7FtFG5Xsc0Sr3d+r0AAAAAsDQxXRUaMgxjfr/Worn1rqtXfuG5Rufr1Q0AeK9DLx7S0fuPKns+K8dylEglFE/GJVMKvECe48m13PkRHEEQ6JGXHmlb/Y06KtvZCRumE7rVOhfeK8xf7y8+VilTrWyjv8hvdLxeAqEZUdvZqFw1rcSzFVFjGrXjvVMxbfTOm41pN36HqrUjSmwAAAAAYCFGcqCuSpKhkmBYnJQIc10QBHUTFJXz1RIcC8/VqhsAUF16V1oPf+th5S7nNPPmzPyUVbmp3PxojvylvLJTWTk5RwdPHNTYjs6vxdFoGqFe/yV6p9TrzO5mXY3qi9rOVursd/Xa36uY9pNWfmcH5RkBAAAA9C9GcqChSvKhUaIh7HXN1gsAiG5i94QeKz2m4w8e1+tffV3x4bgC/1pC2jTkFl1tfXBr6CmqwnQ8tto52c6O0nZqRx3N3qOVOntRttl1NTohauKg1ft2qny3Ytqrsu0oDwAAAOD6RpIDPRd2OuUwlk8AACAASURBVKt6SY+f/d3P9PL/+bLe+vu3NHd+TpK08saV2vihjfrI//wRbfzwxja3GgAGy/5n9mv/M/s1fWpamVcyksojPca2dX7kBgAAAAAAQKeQ5EBP1Zqiqtr5xecqvvbg13T2xFkZcUPL1izTxIcm5Lu+ileKOvPfzujst8/qtv/hNn3qy5/q3IMAwIAY2z6mse0kNgAAAAAAwNJAkgMD7T/9wn9Sbjqn8bvGldqQUjKVlCQ5BUeluZIKVwqyLlv6x7/8R/3xjj/WZ179TI9bDFy/wo7MijK6a/EUeVFHheH60sxC0kynAwAAAABAfyLJgYYqIygarbMR9rpm663lT3f/qazLlib3Tmrt7Wu1YnyFYsMxeSVPpdmSrMuWkqmkYkMxGTFDV398VU999CnpV9rSPABNqPy7UPk3otbvd73rFpepN/Jr8T3DjAoDAAAAAADA4CHJgboWdjRWfq5Y2FHY6LrF+ws7LRfXt7juaude/ZNXdeVHVzS5d1I3fPAGbdi6QStvXClJKr5dVP5iXrFkTDKkwA/kO768kqfpH0xLN0i6M3pMAESz+N+LZq/rZmJi6uSUMievrVuxM63xHeNdqxvdw+gMAAAAAAAGH0kONFSrY3Hx8bDXhT1X83wgvfi/v6jVt6zWyhtXavXm1Vr/gfVae8daWZctZZNZ+Z4vt+jKsRw5eUd2zpadtTU0MiTnbxySHMASVCsh0kyy5NiBYzp9/LQSyxOSIenapY7laMsDW3Tg2QNtbzcAAAAAAACiI8mBgTP781llM1mtvmW1hkeHNbRySPHhuDzbk2EYMhOmYsmYYkPlLT4UV3y4vCWWJ6SfS0a2PVNqAeiNRlNVNTPNlSSdf+m8juw5otS6lDbds0nJkaQM05Dv+LJztoozRZ05cUZPDD2hQy8eUnpXuoNPBwAAAAAAgLBIcqCmoaEhOY4z/3MikZAkua6rUqmkVColScrn813dnzo5JSNmKJaMyTANeY6nwtWCgiAoj+AouPId/52/3jYlwzRkxA2ZcVOmacp7y+tc4AB0VLvX1Dj/0nk9/cmnNb5jXGtvX6vUhpQSyxMK/EBOzlFhplBe42ckqexUVkfvP6qHv/Vw2+oHAAAAAABAdGavG4D+9Gu/9mv6lV/5Ff3whz+c3ypOnz6tL37xi/M/d3t/9mezMmOmAj+QW3RVnCkqdyGnbCar/MW8im8XZedtuQVXnu3Jd30FfjA/7YxpmDJmGckBdFtlyqh663E0uq4Ti4Yf2XNEa25do/Ed40rvTGvjhzdq/K5xrf/Aeq2+dbVGJ0e1cmKlRsZGtGJ8hRLLEnrqnqfa2gYAAAAAAABEYwQdXsn18OHDCoJAn//85ztZDa4jZ/76jL6676tK70pr3R3rNLp5VCMbRpRc8d7pZQpXy3+Bnb+YV+5CTvkLec3+fFbeb3r6/Jc/3+tHAZa8hf8PqLUuRrXppGpdt9jCKamqHW90z2MHjunnL/1cm+/drPTOtMZ2jGl0clSe7ZX/3ZjOKTdVTqIu3GbenJE9aSs4wP/fAAAAAAAAeonpqjBwxu8al+/6Ks2VVLhSKE8r4wVKrkjOj/BwCo7srK3SXEl2zpZjOfJKXnlkh+/LmGAkB9BttXLqi4+HvS7suXrnTx8/rU33bFJqQ0orJlZodHJUqzatUnGmKM/25Fjlf0uSI8nylkoqkUpoePWw7NN23ToBAAAAAADQeSQ5MHCWr1uuDVs2KH8xr/zKvMyEKc/xlJxLykyYUiB5tic7b78r0WHny5vGpGC4owOYAAyAqZNTSixPKDmSVGJ5ovxvie2pOFOUW3QV+OV1fYzYtfV8EqbMpKl4Mq54Mi7FJWOahCkAAAAAAEAvsSYHBtLH//3HZb1tKX8h/66pZHJTOeWmy9NSWZctFa4UVJwplhMdWVtuyZU+0evWA9eXO+64Q1NTU/NbxcJjC4+/8MILXdnPnMxIhmSYRnkEWN5R/mJe2UxW1mVLds6WW3Tfta6PYRiSqXLiwzClTOSwAAAAAAAAoA0YyYGBdOMv3qj373u//vEv/lFBEMgpOkqmkoolY5IhBV4gr+SVp63KlUdzFN8u6oOf/qC+v/H784uQA+isxx9/XJlMpup0UWGmper0vgLJd3w5OUeFqwXFh+NyLEeGYcgtuirNleTkHblFV57jlRMeXlBOegAAAAAAAKDnSHJgYH3qy5/SX/32X+m///l/l2O9k+So/FW27/hyio6cfDnRse23tulX/+hX9f3D3+9104HrSjqdbur43r17u7Kf3lmu387ZKswUNHRpSIZhyM7aMmKGfNeXk3fKI8Hy5bV93pXsCAIFaZIdAAAAAAAAvUSSAwPt1//01zWxa0L/9XP/VYXLBRkxozyaQ5JX8sp/bW1Kn/j3n9D2R7b3uLUA+sn4jnE5lqPiTFHWZUvxobh811dyJCkzbirwA7lFV3beVmm2vLaPYzlyLbe8ZocbSGO9fgoAAAAAAIDrG0kODLy7/qe7dOfBO3XqqVP6yTd/oguvXZAkbfjoBt32idu047d3lBckB4BFtjywRWdOnCknNmLlhceTI8nyvxmB5DmeHOvatHezpflRHcXZovTBXrceAAAAAAAAJDmwJMSH49r1u7u063d39bopAAbIgWcP6ImhJ5SdykqS3KKrZCopM2nKMMpTVrlFV45VnraqNFsqJzh8SfvF+j4AAAAAAAA9xp+3AwCua4dePCQn5yh7PqtsJqu5zJxymZyyU1nlpnPKX8rLumSpcLUg66qlIAj0yEuP9LrZAAAAAAAAEEkOAMB1Lr0rrYe/9bByl3OaeXNG2Uw52ZGbypWTHBfyyl/KKzuVlZNzdPDEQY3tYDEOAAAAAACAfsB0VQCA697E7gk9VnpMxx88rte/+rriw3EFfnkuKsM05BZdbX1wq/Z9ZV+PWwoAAAAAAICFSHIAAHDN/mf2a/8z+zV9alqZVzKSyiM9xrYxcgMAAAAAAKAfkeQAAGCRse1jGttOYgMAAAAAAKDfkeQAAKBFhmHM7wdBEOm6TpwDAAAAAABY6lh4HACAFlSSDJUEw8KkQ9jrDMNQEATzWzvOAQAAAAAAXA8YyQEAQIsqiYtGiYZa19UbgdHu0RlTJ6eUOXltvZGdaY3vGK963eST0e5/7tHGZc89Wv142Dprla91j3rXN6tWG8PWEbV9i8t1u75O1dlqPOvdr9E9mo1p1O+6XvlOvIt2f6NR6mq27k7/3gIAAABY2khyAADQB6JMO9VMsuTYgWM6ffy0EssT0v/P3v3HyHHed57/VE13z2h6hhz+irqbMmkmEmWbpi2NScG3Fy6ZxLAWRhwkJIW1JNwKoLD/BAGE+2MD3N4FJhHdP3cBFgLWduDkRORAS5a41N/G6i4xAR0DOGK0sLQ8RLeMLJvqHpGUNDP9q7qquur+aPZoNJ7uqq6u6q6eeb+Agnqq6qnnqW9VU8Dz7ed5DEn3TnUajo48cURnXz0bqd1RHHwh/g7Mfp2tcdUXVIcUrUO4X/uiJpuGiUeUdkapc9h4DlJ/lPPiFHdMe5UbJqZpisv6YyQ7AAAAAAQhyQEAQAqsT1B0p6HaaOP+oL8l6da1W7p48qLye/M6cOKAcnM5GaYhz/Fk12xZy5Zuvn5Tz08/r3NvnFPpeElScEdomI7Hfp2wm3XShunsDaq3V0d6XImOoLiE6Xxefzxs+7rHBu2IHjQew7Zzszo3Xjfo3EHriyqumAaJGtOo5YaN6SBxGeY7GzTqZhTvAAAAAICtgTU5AACYAL0SH/3cunZLL337JRUXizr0jUPa/9h+lY6VVHikoH1f3KfdD+3WwqEF7Tm8R/n787r0+CV98LMPErqDjiQ6K/t1psZZX5iO4UGvESZhELVTfZh4DNrOoA7tQdsS5z1vdu1xdJoPGtMo5YaJaVriErQfAAAAADYiyQEAwJC6U0YFLfzd67ww5aKszXHx5EXtfnC3iotFlY6V9MDXH1Dxa0Xt+9I+7XpwlxYOLmjH/h2aK8xpvjiv7H1ZvXjixYHrSVpc0+iMYzqe9fUGdeaOun0b60trO8NKY6d41JhO+rNIa7sAAAAAbE1MVwUAwBC662J0ExW9po/qd16/tTU2S4yEWcD88tnLyu/La8/hPdr7hb0qLBa0cHBBbbut+u26MjMZGYYhz/XUtttyLVdOw5G1aum177ym0z8+PXxwYpbGTmyMf+2EcSeHeC97IzYAAAAARoGRHAAADMn3/bVt4/4w5wVdY+MWptyNKze068Fdyt+f1/z+eS0cXNDOAzs1u3dWMwszmt4xrdxc7tMtn1M2n9XMrhm9/erbw4Sjr636C++tel9RHHzhs1vUawxy3jg704e5z1FKWxuDRqOk4dkCAAAAmAyM5AAAYIupXK8oO5tVbi6n7GxWZtZU227LWrbkWq58796okilDZsaUmTVl5kxlcpnONpPR0ltLKjxaGKod/TpVw3ZchunofP+5Tzuaey20PSpp6JAdVzyS6KyOusB9Usb9fsUh6XgNuj5KvyRRGr5PAAAAANKPkRwAAET08MMPq1KprG1d6/f12n/16tW1/XF/Ll8vS4ZkmIZ8z5dTd1S/XVe1XFXjbkN2zZZrufJcT77nS/696bBMyZjqlCm/WR42PGMRxyiCKHVK6eyQHUc8ugtYR13IOmw805DAWr8vzQmPNL+jAAAAADAsRnIAABDBd7/7XZXL5Z5TT22mu3/jQuJJfJYveY4np+ao+XFTmZmMnIYjwzDkWq5aqy05dUeu5arttDsJj7bfSXrEpFeHaq9RBnHU1Wu9hKQ6oNPaeTyuePRKBIR95oMmONIQ9+79pVWaYrVRv7Yl8e8EAAAAgK2JJAcAABGVSqXI+4vF4trnU6dOxfq5dKxTj12z1VxuavrOtAzDkF21ZUx1Fht36o5aqy3ZdVtO47PJDsM0VDq++T3EIWynd5TO2VF2iKa587grzW3bKEo8+yUXJuH5JG3UMRikvqBzB0mOAQAAANjeSHIAALDFFBeLchqOrGVLjbsNZaYz8lxPubmczIwp3/PlWq7suq3WSkt27V6io+HKtTpb4ZHh1uNIkyQ6eie5Az2NbU9jmyYdMQUAAACwXbAmBwAAW9CRJ4501uC401BtqaZqudrZKlXVlmqq36mrcbeh5idNtVZaa6M6rBVLR588Ou7mb6npf4KmiEpLZ3TUdg7b7qgjdnptG89JWr93ddiYRn1nxvFOpfk7CwAAAGBrI8kBAMAWdPbVs3IazlpSo1quqvpBJ9FRW6qp/mFdjTsNNT9qqvlxJ9FhrViSJ53+0elE2zZIZ2jYTtpRJRCGvd7Gdia9Vkiv/YMu6h22nZudF7TuQpj2jFtQPKXRxTSo3LhjOmi9YWILAAAAAP0wXRUAAFvUuTfO6dLjl1S9VZXTcJTNZ5XJZSRT8tu+2k5bbsNdG8Hh+76evfZsbPUHdVLG3QmbdKfo+uuH+fX+xn3dMpuVDfpFftD+fgmEQURtZ1C5zQwTz2FEjWnU9yupmAY980FjOsy7FkXYdybtCTAAAAAA48dIDgAAtqjS8ZKe/snTqt2tafkXy2tTVtUqtbXRHPU7dVUrVTk1R8+8/owKi8mvxRE0jVCcC46PasqiMPq1cZR1BdUXtZ2T8Ayi6tf+ccU0TaKOHukXu63w3gAAAAAYDcP3fT/JCi5cuCDf93X+/PkkqwFC450EsB1defKK3nnlHWVmMvK9zv/6DdOQa7k6+uTRxKeoAgAAAAAASALTVQEAsA2cefmMzrx8RktvLan8ZllSZ6RH4ZHkR24AAAAAAAAkhSQHAADbSOHRggqPktgAAAAAAABbA0kOAACwLQ2ykDRrAwAAAAAAkE4sPA4AAAAAAAAAACYSIzkAAMC2xOgMAAAAAAAmHyM5AAAAAAAAAADARCLJAQAAAAAAAAAAJhJJDgAAAAAAAAAAMJFIcgAAAAAAAAAAgIlEkgMAAAAAAAAAAEwkkhwAAAAAAAAAAGAikeQAAAAAAAAAAAATiSQHAAAAAAAAAACYSCQ5AAAAAAAAAADARCLJAQAAAAAAAAAAJhJJDgAAAAAAAAAAMJFIcgAAAAAAAAAAgIlEkgMAAAAAAAAAAEwkkhwAAAAAAAAAAGAikeQAAAAAAAAAAAATiSQHAAAAAAAAAACYSCQ5AAAAAAAAAADARCLJAQAAAAAAAAAAJlJm3A0AgEEZhrH22ff9SOdFORa2XgAAAAAAAACjwUgOABOlm2joJhnWJx7CnmcYhnzfX9s2HutVrns+AAAAAAAAgHRgJAeAidNNNGxMUIQ9LyhREfb6YVSuV1S+XpYklY6VVFwsbnrewReiXf/954LLvv9c8HU2u0aS5cLqdW9h64jrvkZdX1J1DhvPftcLusagMR32vR7Vs4j7HU36XRu2LAAAAAAAaUOSA8C2lPTUU5fPXtaNKzeUnc1KhqR7VTgNR0eeOKKzr56Nvc5eDr4weKd3ULl+ZYPKhRXUNin+9kVNNo0jjoPWOWw8B6k/ynlxijumvcoNE9Ok3rWodcb1vQUAAAAAYNRIcgDYljautRFXouPWtVu6ePKi8nvzOnDigHJzORmmIc/xZNdsWcuWbr5+U89PP69zb5xT6XhJUnCnZJjOx36dsEEdmL06xMN07q4/Hra+sILiklT7uscG7YgeRxwH6XyPGs84xBXTIFFjGrXcsDEdJC5BI2eS+r4DAAAAAJBmrMkBADG5de2WXvr2SyouFnXoG4e0/7H9Kh0rqfBIQfu+uE+7H9qthUML2nN4j/L353Xp8Uv64GcfJNqmML9aH6TjNui8ODtIB+mkDXtemIRB1E71UcYxKPk1aFvivOfNrj2OjvOo7+Yg5YaJ6TBxGeS5d8XxngIAAAAAkEYkOQBMnO5UU0HrZfQ6L2q5IBdPXtTuB3eruFhU6VhJD3z9ARW/VtS+L+3Trgd3aeHggnbs36G5wpzmi/PK3pfViydeHKiOcdj4C/OwHezjmKZofb1pa9+kxTFIGjvGo8Z00p9FnLbDPQIAAAAAthamqwIwUbqLgXcTEL2mnep3Xr+FyIOuv/Fz9/jls5eV35fXnsN7tPcLe1VYLGjh4ILadlv123VlZjIyDEOe66ltt+VarpyGI2vV0mvfeU2nf3w6ngBhy4uyfkYS9Y+rvjQmVwAAAAAAwPgwkgPAxPF9f23buD/MeVGPrd+/8fiNKze068Fdyt+f1/z+eS0cXNDOAzs1u3dWMwszmt4xrdxc7tMtn1M2n9XMrhm9/erbw4Sjr636q+ytel9RHHzhs1vUawxy3jgTDcPc5ygl0cagESVpeD4AAAAAAIwaIzkAYEiV6xVlZ7PKzeWUnc3KzJpq221Zy5Zcy5Xv3RsdMmXIzJgys6bMnKlMLtPZZjJaemtJhUcLQ7WjX6dqr3n4ux3GvRYiTrs0dOaOK45JdHRHXeA+KZP6Xq4Xd7zWv2+D1LcVvu8AAAAAAGyGJAeAiVGpVD7zd7FYDNyfy+X0zjvv6OTJk5Kkq1evxv75py//VDIkwzTke76cuqP67bradlu+58uu2XItV57ryfd8yb833ZUpGVOdMuU3y0MnOYYxSZ2caf61+jjiOGyHddh4jvre+iXmuu1J4zsgpfsdlSbr+w4AAAAAQBCSHAAmwoULF/TQQw/p1KlTv3Zssymnuvs3TiuVxOfODslzPDk1R82Pm8rMZOQ0HBmGIddy1Vptyak7ci1XbafdSXi0/U7SIya9OlR7/Xp7fZle6x6krTM0rZ3H44rjoCN0Nho0wZGGuK9PdKRR0rHqd/2g5z5p33cAAAAAAMIgyQFgYrz77rt66qmnfm1/qVTa9Pzu/vWJkUQ+P3VK7/3gPdk1W83lpqbvTMswDNlVW8ZUZ7Fxp+6otdqSXbflND6b7DBMQ6Xjm99DHMJ0eqeh8zpImjrae0lz2zaKEs9+neCT8HySNs4ER3d/mATXdn5GAAAAAICthyQHAAypuFiU03BkLVtq3G0oM52R53rKzeVkZkz5ni/XcmXXbbVWWrJr9xIdDVeu1dkKj4xvqqpe0tRpnaa2DCqNbU9jmybdpMd00tsPAAAAANi+zHE3AAC2giNPHFG1XFXjTkO1pZqq5Wpnq1RVW6qpfqeuxt2Gmp801VpprY3qsFYsHX3y6LibP5CgqW3i7iwd9Hqjbl9UUds5bLuj3P/7z/XeNp6TtH6jSYaNadR3Ji3vFAAAAAAA2xFJDgCIwdlXz8ppOGtJjWq5quoHnURHbamm+od1Ne401PyoqebHnUSHtWJJnnT6R6cTbVuYKYZ67R90Meq45/QftvM46fYFXTfpOG52XtCaDWHaM25B8ZRGF9OgcuOIaZj4RCmX9vcCAAAAAIDNMF0VAMTk3BvndOnxS6reqsppOMrms8rkMpIp+W1fbactt+GujeDwfV/PXns2tvqDOjiDfoU+iPWLP29WPo7O0vXXDfPr/Y37orQvbOdxvwTCIKK2M6jcZoaJ5zCixjRqQiqpmAY980FjGiUuYZ97v2fI4uIAAAAAgK2GkRwAEJPS8ZKe/snTqt2tafkXy2tTVtUqtbXRHPU7dVUrVTk1R8+8/owKi8mvxdFvGqF++4M6u9O+iPko2zeOOA5TZ9oFLZo9jpimRdD3OcqC41vhnQEAAAAAbF+G7/t+khVcuHBBvu/r/PnzSVYDhMY7OZkm7bldefKK3nnlHWVmMvK9zj+zhmnItVwdffJo4lNUAQAAAAAAANsB01UBQALOvHxGZ14+o6W3llR+syypM9Kj8EjyIzcAAAAAAACA7YIkBwAkqPBoQYVHSWwAAAAAAAAASSDJAWxRhmGsfe43K12v8/qVD3ssqG5sL4MseMz6AAAAAAAAAAiDhceBLaibaOgmGDYmHsKc5/t+zwRFv2Prj5PgAAAAAAAAAJAkRnIAW1Q3weD7fs8kxyDnJalyvaLy9XvrVhwrqbhYHEs7kCxGZwAAAAAAACBuJDkAxC7sVFmXz17WjSs3lJ3NSoake6c6DUdHnjiis6+eTbilAAAAAAAAACYZSQ4AsdpsjY6N+25du6WLJy8qvzevAycOKDeXk2Ea8hxPds2WtWzp5us39fz08zr3xjmVjpdGeQsAAAAAAAAAJgRJDgAjdevaLb307ZdUXCxqz+E9yt+fV3Y2K9/z5dQcNZebatxtKDeXU7VS1aXHL+npnzw97mYDAAAAAAAASCGSHMAW1R1BEbTORtjz4nLx5EUVF4sqLha19wt7Nb9/XmbWlFN31Py4qek708pMZ2ROmZKk6q2qXjzxovTvR9I8AAAAAAAAABOEJAewBXWTFt3ExfrpotZPHxV03sbP3eNhj2285uWzl5Xfl9eew3u09wt7VVgsaOHggtp2W/XbdWVmMjIMQ57rqW235VqunIYja9WSfdmWWKIDAAAAAAAAwDokOYAtqteC3xv3hz0vjmM3rtzQgRMHlL8/r/n981o4uKCdB3bKWrbUtttyGo7sqq3cXK6z5XPK5rOa2TUj+4bd87oAAAAAAAAAtidz3A0AsD1UrleUnc0qN5dTdjYrM2uqbbdlLVtyLVe+d29UyZQhM2PKzJoyc6YyuYwyuYyUkYyl0UypBQAAAAAAAGAykOQAtphKpfKZTZJ+/vOfrx2/evXqWD7/9OWfSoZkmEZnkfG6o/rtuqrlqhp3G7JrtlzLled68j1f8u9NfWWqk/gwTKk8cDgAAAAAAAAAbGFMVwVsIRcuXNBDDz2kU6dOfWb/+imkxvW5s0PyHE9OrbPIeGYmI6fhyDAMuZar1mpLTt2Ra7lqO+1OwqPtd5IeAAAAAAAAALABIzmALebdd99VqVRa2yTpq1/96trx9QmQkX5+qvPZrtlqLjfVuNNQrVJT9YOqqpWq6nfqan7cVGu1Jbtuy2lsSHb4vvwSyQ4AAAAAAAAAn2IkB4CRKC4W5TQcWcuWGncbykxn5LmecnM5mRlTvufLtVzZdVutlZbs2r1ER8PtrNnh+lJh3HcBAAAAAAAAIE1IcgAYmSNPHNHN1292EhtTnYXHc3M5mVlT8qW205bTcGTXOomO7qgOa8WSvjzu1gMAAAAAAABIG5IcAEbm7Ktn9fz086pWqpIk13KVy+dk5kwZhiHP9eRarpyGo9ZqS62VVifB4Uk6I4nZqgAAAAAAAACsw5ocAEbq3Bvn5NQcVW9VVS1XtVpeVa1cU7VSVW2ppvqduhp3Gmp+3FTj44Z839ez154dd7MBAAAAAAAApBBJDgAjVTpe0tM/eVq1uzUt/2JZ1XIn2VGr1DpJjg/rqt+pq1qpyqk5eub1Z1RYZDEOAAAAAAAAAL+O6aoAjNz+x/brz1p/pitPXtE7r7yjzExGvteZi8owDbmWq6NPHtXpH50ec0sBAAAAAAAApBlJDgBjc+blMzrz8hktvbWk8ptlSZ2RHoVHGLkBAAAAAAAAIBhJDgBjV3i0oMKjJDYAAAAAAAAADIYkB4CJYxjG2mff9yOdF+VY2HoBAAAAAAAAjAYLjwOYKN1EQzfJsD7xEPY8wzDk+/7aFvZYdx8AAAAAAACAdGAkB4CJ0000bExChD2vX6Ii7iRG5XpF5ev31hs5VlJxsbjpeQdfiHb9958LLvv+c72P9Srbr0y/8mHKDdPesOK+r7Blo8QjzvqSrHPYmPa6VlD5QWM67Ps1qmcxju9eUP1JxQYAAAAAgCSR5ACwLUWdyiqsy2cv68aVG8rOZiVD0r3LOA1HR544orOvno103SgOvrB5R2S/zuDusV4dmFGTMqMwzH31K98rjkF19hPU1rjbGbXOYWMa9lpRzotT3DFN43dv3M8fAAAAAIC4keQAsC1tXGtj/d/9jgW5de2WLp68qPzevA6cOKDcXE6GachzPNk1W9aypZuv39Tz08/r3BvnVDpekhTcoRmmA7Ff52S/Ttqo5daXjdLxmmSnaFA8E00c8gAAIABJREFUw3Tmrj+edDw2XjPpdm5W58brhjl/0DqjiCumQaLGNGq5YeM5zHevV/2bXSto1E3Szx8AAAAAgCCsyQEAMbl17ZZe+vZLKi4Wdegbh7T/sf0qHSup8EhB+764T7sf2q2FQwvac3iP8vfndenxS/rgZx8k2qagTtJBEh9hy45bmI7hQa+RRDz6JbCSamdQ0izK+zDIfQ+atBvHOzZoTKOUGyaew8Ql6vOPWgYAAAAAgFEgyQFg4nSnk+q3Hke/8/qVC7pmPxdPXtTuB3eruFhU6VhJD3z9ARW/VtS+L+3Trgd3aeHggnbs36G5wpzmi/PK3pfViydejFwf4hW2A3jU0yhtrC+t7QwrjZ3iUWM66c8CAAAAAICtgOmqAEyU7iLi3WREr6ml+p3XbyHyfsfW79943ctnLyu/L689h/do7xf2qrBY0MLBBbXttuq368rMZGQYhjzXU9tuy7VcOQ1H1qql177zmk7/+PTwwZkwwy6cjMGMe+2EcSeHtvv7Ne7nDwAAAABAUkhyAJg4vdbI2Li/31oaUY71K3Pjyg0dOHFA+fvzmt8/r4WDC9p5YKesZUttuy2n4ciu2srN5TpbPqdsPquZXTN6+9W3E0tyDLsg9ig7REdRJ7+o/1RcSYAwMU1DB3sa2hDGqN7RsM///ec65watSZL2uAIAAAAAti6SHAAwpMr1irKzWeXmcsrOZmVmTbXttqxlS67lyvfujSqZMmRmTJlZU2bOVCaX6WwzGS29taTCo4Wh2hFl0eh+ZUe5MPiokw9p6JDt13mcZDx6XXvYzuooC50nZdzvVxySileU57/+Xd1MGr5PAAAAAIDtizU5AEyMhx9+WJVKZW3rWr+v1/5/+Id/WNt/9erVWD+Xr5clQzJMQ77ny6k7qt+uq1quqnG3Ibtmy7Vcea4n3/Ml/950V6ZkTHXKlN8sDxue2CXVMRy00HZS9ab5F+fdDuR+Hclx6y5gneRC1uvPGZVxvV9xGOU7GsfzBwAAAAAgDRjJAWAifPe731W5XN50yqgw00t5nrfp/rg+y5c8x5NTc9T8uKnMTEZOw5FhGHItV63Vlpy6I9dy1XbanYRH2+8kPWLSq6Oy31Qz/cqGKRen7q/Fk5DWBEevhamTXrA66Nf6YeI0SIIjDXFP8v2Kw6gTHJvtCxpZlIZ/JwAAAAAA2IgkB4CJUSqVIu9f//nUqVOxfi4d61zbrtlqLjc1fWdahmHIrtoypjqLjTt1R63Vluy6Lafx2WSHYRoqHd/8HuIwaOf1sOXSJk0d7b2kuW2bGTSm/ZILk/B8kpbmGAS1bav8OwEAAAAAmFwkOQBgSMXFopyGI2vZUuNuQ5npjDzXU24uJzNjyvd8uZYru26rtdKSXbuX6Gi4cq3OVnhkuPU4sLk0dx4HSWvb09quSUU8AQAAAAAYDmtyAEAMjjxxpLMGx52Gaks1VcvVzlapqrZUU/1OXY27DTU/aaq10lob1WGtWDr65NFxNz8V4p5KaNDO46ApotLSGR21nXG0O0pMe20bz0lav/dr2JhGfWfGPUUVAAAAAABbAUkOAIjB2VfPymk4a0mNarmq6gedREdtqab6h3U17jTU/Kip5sedRIe1YkmedPpHpxNtW1AH7KDlkm7LODrj+7Vns7/jMmxCJWo7NzsvTR3yUcXxfsUV07Dfr3HEc5jnH3Y/AAAAAACjwnRVABCTc2+c06XHL6l6qyqn4SibzyqTy0im5Ld9tZ223Ia7NoLD9309e+3Z2OoP6mzstWhwkEHK9VpAe9A6h7H++mF+vb9xX7fMZmWH7QCOKx5R2xlUrpdhYhpV1JhGfb+SimnQMx80nsO8a1Gef9gyaU5+AQAAAAC2NkZyAEBMSsdLevonT6t2t6blXyyvTVlVq9TWRnPU79RVrVTl1Bw98/ozKiwmvxZHr+mAgjolk5hGqN/1RjVtUZB+ndmjrCvM8xlkfxx1pt2w71cSMU2bKM8/6jEAAAAAAEbB8H3fT7KCCxcuyPd9nT9/PslqgNC28ju5le9t0lx58oreeeUdZWYy8r3OP7OGaci1XB198mjiU1QBAAAAAAAA2wHTVQFAAs68fEZnXj6jpbeWVH6zLKkz0qPwSPIjNwAAAAAAAIDtgiQHACSo8GhBhUdJbAAAAAAAAABJIMkBAEi1QRaSZm0AAAAAAACA7YWFxwEAAAAAAAAAwERiJAcAINUYnQEAAAAAAIBeGMkBAAAAAAAAAAAmEkkOAAAAAAAAAAAwkUhyAAAAAAAAAACAiUSSAwAAAAAAAAAATCSSHAAAAAAAAAAAYCKR5AAAAAAAAAAAABOJJAcAAAAAAAAAAJhIJDkAAAAAAAAAAMBEIskBAAAAAAAAAAAmEkkOAAAAAAAAAAAwkUhyAAAAAAAAAACAiUSSAwAAAAAAAAAATCSSHAAAAAAAAAAAYCKR5AAAAAAAAAAAABOJJAcAAAAAAAAAAJhIJDkAAAAAAAAAAMBEIskBAAAAAAAAAAAmEkkOAAAAAAAAAAAwkTLjbgAAYOswDGPts+/7kc7rdWz9/qDj/eoGAAAAAADA1sFIDgBALLpJhm6CYbOkRNB5hmHI9/21bWPiYv228Zq9ygEAAAAAAGDrYiQHACA23eRDUKKh13lhR2B0kxrDqFyvqHy9LEkqHSupuFjc9LyDL0S7/vvPBZd9/7ng62x2jSTLhdXr3sLWEdd9jbq+pOocNp79rhd0jUFjOux7PapnEfc7mvS7NmxZAAAAANiuSHIAAFIlStJjkGTJ5bOXdePKDWVns5Ih6d6pTsPRkSeO6OyrZyO1O4qDLwze6b3+2GZlg8rF0WEatW1B5aPGo59h4hGlnVHqHDaeg9Qf5bw4xR3TXuWGiWlS71rUOqO8AwAAAACwnZDkAACkysZ1NnpNTdVv32bn3Lp2SxdPXlR+b14HThxQbi4nwzTkOZ7smi1r2dLN12/q+ennde6NcyodL0kK7pQM0/HYrxM2qNN70LK9yoUpM4iguITpfF5/PGz7uscG7YgeNB7DtnOzOjdeN+jcQeuLKq6YBoka06jlho3pIHEZ5jsbx/cdAAAAALYz1uQAAGx5t67d0kvffknFxaIOfeOQ9j+2X6VjJRUeKWjfF/dp90O7tXBoQXsO71H+/rwuPX5JH/zsg0TbFDU50m9/v8RLnJ2jYTqGB71GmIRB1E71YeIxaDuDkl+DtiXOe97s2uPoNB80plHKDRPTYeIy6Hc2rrIAAAAAsJ2R5AAAxKY7ZVTQwt+9zktqwfCLJy9q94O7VVwsqnSspAe+/oCKXytq35f2adeDu7RwcEE79u/QXGFO88V5Ze/L6sUTLybSljQYxzRF6+sN6swddfs21pfWdoaVxk7xqDGd9GcBAAAAAEge01UBAGLRXRejm6joNX1Uv/OC1tboteB4v3KXz15Wfl9eew7v0d4v7FVhsaCFgwtq223Vb9eVmcnIMAx5rqe23ZZruXIajqxVS6995zWd/vHpYcKCbWTcayeMOzmUxuQKAAAAAGDrYyQHACA2vu+vbRv3hzkvzLFB675x5YZ2PbhL+fvzmt8/r4WDC9p5YKdm985qZmFG0zumlZvLfbrlc8rms5rZNaO3X317kNsfSL8O6aBfp4+7M70fflH/qYMvfHaLeo1BzhvnOzHMfY5SEm0c5js7yd93AAAAAEgDRnIAALasyvWKsrNZ5eZyys5mZWZNte22rGVLruXK9+6NKpkyZGZMmVlTZs5UJpfpbDMZLb21pMKjhaHaESahsdn+fp3GvTpLu2X6LW48CmnokB1XPJLorB5mTYckjPv9ikPc8YrynY2jLAAAAABsdyQ5AABDq1Qqn/m7WCwOvP/nP/+5vvKVr0iSrl69qpMnTw79+acv/1QyJMM05Hu+nLqj+u262nZbvufLrtlyLVee68n3fMm/ty6IKRlTnTLlN8tDJznGYRydzmn+xfk44jFsIiBsPNOQwOp20nfbk8Z3QEr3OwoAAAAAiIYkBwBgKBcuXNBDDz2kU6dO/dqxXtNLBU1FFdfnzg7Jczw5NUfNj5vKzGTkNBwZhiHXctVabcmpO3ItV22n3Ul4tP1O0iMmvTpUe40y6B7rVbZfuV5T3yS9QHNaO4/HFY9BR9psNGiCIw1xX5/oSKOkYxX1OztsWQAAAADY7khyAACG9u677+qpp576tf2lUmnT8zfb/9WvfnXt8/qEyVCfnzql937wnuyareZyU9N3pmUYhuyqLWOqs9i4U3fUWm3JrttyGp9NdhimodLxze8hDr06vYM6Y8N0lo+yQzRNHe29pLltG0WJZ7/kwiQ8n6SNM8HR3R80ddow33cAAAAA2M5IcgAAtqziYlFOw5G1bKlxt6HMdEae6yk3l5OZMeV7vlzLlV231Vppya7dS3Q0XLlWZys8MnlTVfWSREfvJHegp7HtaWzTpCOmAAAAALC1meNuAAAASTryxBFVy1U17jRUW6qpWq52tkpVtaWa6nfqatxtqPlJU62V1tqoDmvF0tEnj467+ak2aOdx0BRRaemMjtrOYdsd5f7ff673tvGcpPUbTTJsTKO+M2l5pwAAAAAAySHJAQDY0s6+elZOw1lLalTLVVU/6CQ6aks11T+sq3GnoeZHTTU/7iQ6rBVL8qTTPzqdaNuC1i8I6tiNUiauzt5hr7exnUmvFdJr/6CLeodt52bnBa27EKY94xbmnRxVTMN+f8YxdVvY/XGVBQAAAIDtjOmqAABb3rk3zunS45dUvVWV03CUzWeVyWUkU/LbvtpOW27DXRvB4fu+nr32bGz1B3VSbvbr9W6ZML+OH7S+Ya2//qDtC7q3Qe+p14LiYcr2E7WdYZ9dr/ZFed5RRY1p1PcrqZgGPfNBYxolLsN8Z4f9vgMAAADAdkeSAwCw5ZWOl/T0T57WiydelLVqaWbXjDK5jIwpQ77ny3M9uZa7NoLj2WvPqrCY/Foc/Tot+03TE1Ru0DKjNso2DlNX1LKT8Ayi6nVv3WNRy2+FmEb9zg5bFgAAAAC2O8P3fT/JCi5cuCDf93X+/PkkqwFC28rv5Fa+N6TXpL13V568ondeeUeZmYx8r/O/QMM05Fqujj55NPEpqgAAAAAAABAfRnIAALaVMy+f0ZmXz2jprSWV3yxL6oz0KDyS/MgNAAAAAAAAxIskBwBgWyo8WlDhURIbAAAAAAAAk4wkBwCkiGEYa5/7zSbY67yg8mGvj+1lkIWkWR8AAAAAAACkiTnuBgAAOroJiG7yYX1CIux5vu/3TF4YhrF23Pf9ntcHAAAAAAAAJgUjOQAgRboJiqAkRNjzhlW5XlH5+r11K46VVFwsJlYXxofRGQAAAAAAYFKR5ACAbWJjQqTfdFWXz17WjSs3lJ3NSoake6c6DUdHnjiis6+eTbi1AAAAAAAAQDCSHACwTXSnq+r1tyTdunZLF09eVH5vXgdOHFBuLifDNOQ5nuyaLWvZ0s3Xb+r56ed17o1zKh0vjfo2AAAAAAAAgDUkOQAAkjoJjpe+/ZKKi0XtObxH+fvzys5m5Xu+nJqj5nJTjbsN5eZyqlaquvT4JT39k6fH3WwAAAAAAABsYyQ5ACBFuqMrgtbZCHveIC6evKjiYlHFxaL2fmGv5vfPy8yacuqOmh83NX1nWpnpjMwpU5JUvVXViydelP59bE0AAAAAAAAABkKSAwBSopu06CYuek0tFXTexs+9FilfX+7y2cvK78trz+E92vuFvSosFrRwcEFtu6367boyMxkZhiHP9dS223ItV07DkbVqyb5sSyzRAQAAAAAAgDEgyQEAKdJrMfCN+8OeF/b4jSs3dODEAeXvz2t+/7wWDi5o54GdspYtte22nIYju2orN5frbPmcsvmsZnbNyL5hh7gzAAAAAAAAIH7muBsAABivyvWKsrNZ5eZyys5mZWZNte22rGVLruXK9+6NHJkyZGZMmVlTZs5UJpdRJpeRMpKxFN+0WQAAAAAAAEBYJDkAIAUqlcpnto37//Ef/3Ft39WrV2P9XL5elgzJMI3OIuN1R/XbdVXLVTXuNmTXbLmWK8/15Hu+5N+bCstUJ/FhmFI5pkAAAAAAAAAAA2C6KgAYswsXLuihhx7SqVOnfu3Y+vU0Nu6L87N8yXM8ObXOIuOZmYychiPDMORarlqrLTl1R67lqu20OwmPtt9JegAAAAAAAABjQpIDAFLg3Xff1VNPPfVr+0ul0mf+K+kzyZA4PpeOda5t12w1l5uavjMtwzBkV20ZU53Fxp26o9ZqS3bdltPYkOzwffklkh0AAAAAAAAYPZIcALDNFReLchqOrGVLjbsNZaYz8lxPubmczIwp3/PlWq7suq3WSkt27V6io+F21uxwfakw7rsAAAAAAADAdkSSAwCgI08c0c3Xb3YSG1OdhcdzczmZWVPypbbTltNwZNc6iY7uqA5rxZK+PO7WAwAAAAAAYLsiyQEA0NlXz+r56edVrVQlSa7lKpfPycyZMozOlFWu5cppdKataq20OgkOT9IZScxWBQAAAAAAgDEwx90AAEA6nHvjnJyao+qtqqrlqlbLq6qVa6pWqqot1VS/U1fjTkPNj5tqfNyQ7/t69tqz4242AAAAAAAAtjGSHAAASVLpeElP/+Rp1e7WtPyLZVXLnWRHrVLrJDk+rKt+p65qpSqn5uiZ159RYZHFOAAAAAAAADA+TFcFAFiz/7H9+rPWn+nKk1f0zivvKDOTke915qIyTEOu5erok0d1+kenx9xSAAAAAAAAgCQHAGATZ14+ozMvn9HSW0sqv1mW1BnpUXiEkRsAAAAAAABID5IcAICeCo8WVHiUxAYAAAAAAADSiSQHACA2hmGsffZ9P9J5SRwDAAAAAADA1sTC4wCAWHSTDN0Ew/qkQ9jzDMOQ7/trWxzHAAAAAAAAsHUxkgMAEJtu4iIo0dDrvH4jMOIenVG5XlH5+r31Ro6VVFwsbnrewReiXf/954LLvv/cYNdcf71BykYpt1nbB21v2OsOcu2o7dpYbtT1JVnnsDHtda2g8oPGdNjvw6ieRdzvaNQ2DlPnIGUBAAAAYNKR5AAATIxeCZFBkiWXz17WjSs3lJ3NSoake6c6DUdHnjiis6+ejb3dvRx8YbhOzCTK9Tt/kPZGubYUrfO5X7uSiFtQHKK0M2qdw8Y07LWinBenuGOaRDyTetei1jns9xUAAAAAJgVJDgBAKnWnoFpv/d/rj288d7Oyt67d0sWTF5Xfm9eBEweUm8vJMA15jie7ZstatnTz9Zt6fvp5nXvjnErHS5KCOxfDdCL260xNa0fkoB3Aw1x3/bXDdD6vPx42jt1jg97Dxmsm3c7N6tx43TDnD1pnFHHFNEjUmEYtN2w844zLoHUOWg4AAAAAtgLW5AAApM5mSYph3Lp2Sy99+yUVF4s69I1D2v/YfpWOlVR4pKB9X9yn3Q/t1sKhBe05vEf5+/O69PglffCzD2KrfzNRppuK2jk86C/5e50/bGfpIJ20Yc8LkzCIM25JtTNq7OOIaZj6N153HB3ng8Y0Srlh4hk1Lv3KhRmlMsx7CgAAAABbAUkOAEBsulNGBS383e+8XgmOYRYTv3jyonY/uFvFxaJKx0p64OsPqPi1ovZ9aZ92PbhLCwcXtGP/Ds0V5jRfnFf2vqxePPFi5PqSELXTcit0doZNAIx6GqWN9aW1nWGl8V2JGtNJfxZx2g73CAAAAGB7Y7oqAEAsuutidJMRvaaPCjpv/X/XH++37ka/Y5fPXlZ+X157Du/R3i/sVWGxoIWDC2rbbdVv15WZycgwDHmup7bdlmu5chqOrFVLr33nNZ3+8enhgzOEUa/FkcaO7q1u3LEfd3KIdw4AAAAAMAySHACA2PSaYqrf2hph9oc53uvYjSs3dODEAeXvz2t+/7wWDi5o54GdspYtte22nIYju2orN5frbPmcsvmsZnbN6O1X304syRGmY3lU01T1u0ZX0h3R/Nr8U3HFPsl3LE5paEMY41xwPe2xAQAAAIBxIskBANiyKtcrys5mlZvLKTublZk11bbbspYtuZYr37s3qmTKkJkxZWZNmTlTmVyms81ktPTWkgqPFoZqR5RFo8Mej7tcr7aOqrM1DZ257z/Xud/NFm1OsqM7qdgn9Y5FMcp4JiXJeG0Wj35Tbo3jPQUAAACAtGFNDgDA0B5++GFVKpW1rWv9vu7+q1evrh1P+vNPX/6pZEiGacj3fDl1R/XbdVXLVTXuNmTXbLmWK8/15Hu+5N+bKsuUjKlOmfKb5ahhGcqopqnqpbsY8igWmU7zr9W7ncjdbRTiiH2YmI66Izxogew0d8yP8x0NOyJn1O8pAAAAAKQFIzkAAEP57ne/q3K5vOl0UUH7kv7c2SF5jien5qj5cVOZmYychiPDMORarlqrLTl1R67lqu20OwmPtt9JesSkV8do0K+wxzFNVa/y/X41Poy0Jjh6LUyd9ILVccR+kARHGuLevb+0GmWsNquj37Mf13sKAAAAAGlCkgMAMLRSqRR6/6lTp0b3+alTeu8H78mu2WouNzV9Z1qGYciu2jKmOouNO3VHrdWW7Lotp/HZZIdhGiod3/ze4hDUed2vg7Jfx2vUcqOWprb0kua2bWbQmE7KuzIuaYhBmCTXdn5GAAAAAECSAwCwZRUXi3IajqxlS427DWWmM/JcT7m5nMyMKd/z5Vqu7Lqt1kpLdu1eoqPhyrU6W+GR4dbjwObS0HkcVVrbntZ2TapJj+ektx8AAAAAwiLJAQDY0o48cUQ3X7/ZSWxMdRYez83lZGZNyZfaTltOw5Fd6yQ6uqM6rBVLR588OpY2R51maJjpiUY5ZdCgna9hR7yMuzM3ajvjiH2UmMZ1rWH1u/dhYxr1nUnLOwUAAAAACMbC4wCALe3sq2flNBxVK1XVlmqqlquqflBVtdz5u/5hXY07DTU/aqr5cVOtlZasFUvypNM/Op1o29I6X/5m7Yqr03fY62xsW1Ix7HXdsO2P2s4osZ+EDvmgeEqji2lQuTQleYKOD/ueAgAAAMBWwEgOAMCWd+6Nc7r0+CVVb1XlNBxl81llchnJlPy2r7bTlttw10Zw+L6vZ689G1v9QZ2YaemIXD+iIInkwfprBv16f7N9/doWlAAI2j/o2ia9RG1n1NgPE9OoosY06juVVEyDnvmg8Uz6XRv0HQcAAACA7YKRHACALa90vKSnf/K0andrWv7Fcmc0R7mqWqW2NpqjfqeuaqUqp+bomdefUWEx+bU43n8uPQmOrn4dxuNu6ygXXR4mDlHbmebYD6tf+8cV07QIcw+D3sdWeGcAAAAAICzD930/yQouXLgg3/d1/vz5JKsBQlv/ThqGsba/31eh13lB5aOWi4rv22TiuY3WlSev6J1X3lFmJiPf63z/DNOQa7k6+uTRxKeoAgAAAAAAQHyYrgrbVjfR4Pu+DMOQYRh9ExWbndf97/qkxfpyGxMbYcoBSNaZl8/ozMtntPTWkspvliV1RnoUHkl+5AYAAAAAAADiRZID29r6pEO/hEPY85LWttuSpKnc1NjaAGwVhUcLKjxKYgMAAAAAAGCSkeQAUu69v31Pf/8Xf6/y9bIadxuSpNl9syp9raR/8af/Qp8/+fnxNhDAtjXIgsesDwAAAAAAAJJAkgNISByjPn78hz/WL9/4paZyU5r9jVnt+q1d8hxP1ieWbv39Lb3yR6/oN3/3N/XEf3oiplYDAAAAAAAAwOQgyQEkqNdi44HlPF/f+9L3ZH1iaf+x/coX8srlc5IvOU1HrdWWmh81Vb9b13/7z/9N3//y9/XHb/9xErcAAD0xOgMAAAAAAIwbSQ5sa93FwIMSEGHPCyof1g+/9kM5NUef/53Pa8/hPZovzmtqekrtVlvWiqXG3Yay+aympqdkTpla/sWy/uqxv5J+P1LzAAAAAAAAAGAikeTAttVNWnQTFxtHXWxcbLzXeRs/d4+vP7YxwdGv3D987x/0yXuf6NCpQ/qNI7+h+79yv3Z8bofkS81Pmqp/WNdUbqrTxravttOW23J15/+9IxUlLQ4XFwAAAAAAAACYFCQ5sK31Gl2xcX/Y84Y95nu+rv3v17T7t3Zrx+d2aOHQgvZ+ca/2HN6jxt2GzKwpz/Xktlw5dUd2ze5s87am56blvOHIWBxuHZBJ0C+BFOa8oPLDHgcAAAAAAAAwGua4GwDgU6u/WlXtw5pm985qeue0puenlZnOqN1qS4ZkZkxN5aY62/SUMjOZtS07m5WqklbHfRfJ6jdiJux5vu/3TE50R/F0t43X33gcAAAAAAAAwPiQ5MC2NDs7K9u217au7t+O42hlZWVt/6g+l98sy5gyZGZNGaahtt1W4+OGquWqrE8suU1XnuPJ9+51rhuSYRqdMhlTpmnK/2Drd7yvn0osjvM2K7OZQddW6fro3Y/00bsfDVwOAAAAAAAAQH8kObDt/OEf/qF+7/d+T//0T/+0tnV1//7Vr36lixcvru0f1efVD1ZlTpny277cpitr2VJ9qa5quar67bqay03ZNVuu1Ul2eO69hMe9fnfTMGWsbv3pqpLWXYNls6TG+mP9vPVXb+l7X/qe/jz35/rh4g/1w8Uf6s9zf67vf+n7+i8v/pckmw8AAAAAAABsG4af8HwrFy5ckO/7On/+fJLVAFvCzf98U6/80SsqHS9p78N7tfD5BeXvz2t6flrGVGdkh1231VpuqfFRQ427DTVuN1S7XVP9w7qWf7Us7zuezv+f58d9K4nZmHjoNboizHlhRmasPyfMNd2Wq79+7K+1cmtF9+26T3O/MSczZ8pzPNk1W/U7dbkNV7t+a5f+7c/+rYwpg38nAQAAAAAAgIhYeBxIkdKxkjzXU2ulk8TI3JeR7/myq7bMjCmv7cltup3Fxqu2nLojp+nItVy1W235bV/GfkZyjEur2tL3Hv6epnJTOvjbBzVzM9k8AAAgAElEQVRXmFM2n5XvfToyJ78vr/rduj557xP9hwP/QX/y7p+Mu9kAAAAAAADAxCLJAaTIfbvv0/1fuV+1Sk3TO6Y1lZmS53jKzeVkZk35nq+23ZbTcNRabam12pJds+U0HDl1RypK/vTWX5OjO4IiaMqosOdtViaKH3z5B8rms3rg6w9oz8N7NF+a11RuSq7VSXA07jaUnc3KzJkyDEMrv1rRXx79S+mZSNUBAAAAAAAA2x5JDiBlvvW9b+lvfudvlLudk4zO9EfZfFZTuSlJkufeG81RtzuJjpWW7Kott+VK/3rMjR+BbtKim7joNX1U0HkbP28st76+jXVvduxv/5e/VWu1pd/8xm9q35F9Kny1oB2f2yHf89X8qKna7ZrMjCn5nWfYtttyW65W3l+Rfirp5LCRAQAAAAAAALYfkhxAyux/bL++/OSX9V9//F/XpjnqJjkMw5DXvtdB3uhMW2WtWrI+sfTVZ76qNwtvri1CvpX1GmmxcX/Y8wY5vtkxz/X05l++qT2H92h+/7wWDi5oz8N7tOs3d6n5cVOGaaw9M6fuyK7aalVbys3lNL1zWs7PHBn/kmnGAAAAAAAAgEGR5ABS6A/++g+Uy+f0j//HP8ppOGtJDtO8N2WV05ZruXIajuyarWN/fEzf/Itv6s0Lb4676dvSJ//8idyGq5ldM5qen1Z2NivDMOQ0HPmeL8M0ZGZNmVlTU7kpTU1PKTOdUWY6o+x9Wemu5H+yDbJTAAAAAAAAQMzMcTcAwOb+1Qv/Sr//g99Xa7Wl1V+u6pN//kTLv1zWyq9WtPLLFdU/rKtttfUHf/0H+uZffHPczR2JarX6ma2rXC6vfX7//fdH/rlyvSIZ0lS2M6WY23LV+Kiharmq5kdNOXVHbbst3/M7I0EMyTANGVOGzIwp0zClT28BAAAAAAAAQEgkOYAU+8r/8BX96d0/1be+/y0d/tZh5ffllf+NvA5/+7C+9f1v6d999O/05Se/PO5mjsTf/M3f6K233lKlUlnbun7+85+vff67v/u7kX9u3G3IMA15rien6cj6xFJtqaZquar67bqanzQ766Y0XbXttjzHk9f2JK8z/ZVpmDIaTFcFAAAAAAAADMrwgyanH9KFCxfk+77Onz+fZDUAtLW/b2m+t/evvq8ffetHKn6tqL2H92rn53dq7v455eZzMqdMte12Z6H4lZaaHzfVuNtQ/U5d9Q872/KtZXlPezp/8fy4bwUAAAAAAACYKKzJAQBDKn6tqLbdVmulpcZHDWXuy8j3fOWqOZkZU37bl2u5alVbaq22ZNdsOQ1HruXKbbny276M/YzkAAAAAAAAAAZFkgMAhpSby+lz/93n9NH/95Gm56dlZky1nbZy+ZzMrCn5Uttuy2k4nUTHSkt21ZZdt2XXbOmA5GdYeBwAAAAAAAAYFEkOAIjBH136I/3Hw/9Rtds1yegsPp7L5zSV6yxG7rmeXMvtTFu1+mmiw7Vc6cyYGw8AAAAAAABMKBYeB4AY7DywU//yu/9StUpN1UpV1fK6rVJVbamm+u26Gncban7cVHO5qVa1pd/9X39Xmh9360fDMIy1Lep5UY6FrRcAAAAAAACTh5EcABCTE//TCWVnsvrb//lvZVdtTc9Pa2p6SoZpyPd9ec690RxVW47l6Jv/2zd1/E+O6/ULr4+76YnrJhh8319LOPj+r0/R1e+8jWXCHlt/DgAAAAAAALYWkhwAEKOv/49f14H//oAu/+vLqi3V1Hbbmsp2pqxq221N5aaU35vXv/m//o0Ki4Uxt3a01icd+iUcep23WVIkKZXrFZWvlyVJpWMlFReLm5538IVo13//ueCy7z8XfJ3NrtGvXK86w9Q1iGHrGfS+epUbdX1J1Rn3c1t/vaBrDBrTYd/rUT2LuN/RJN+1UX1vAQAAAGBSkeQAgJiVHivpuX9+Tr/8f36p9/7v9/Sra7+SJH3utz+nQ79zSAd++8CYW4heLp+9rBtXbig7m5UMSffyKk7D0ZEnjujsq2dH1paDLwze6b3+2MayUcpEMWw9vcpHjUc/QW2Nu51R6oz7uYWNVdSYDiPumPYqN0xMk3rXetU5qu8tAAAAAEwykhwAkARDOvDbB0hoJGTj9FRBo0OC3Lp2SxdPXlR+b14HThxQbi4nwzTkOZ7smi1r2dLN12/q+ennde6NcyodL0kK7ggN0/nYr2MzqNN70LJRywwqKC5hOp/XHw/bvu6xQTuie8U4qXZuVufG6wadO2h9UcUV0yBRYxq13LAxHSQuSXxnB2krAAAAAGx1LDwOAJgovdbz8H1/bRvErWu39NK3X1JxsahD3zik/Y/tV+lYSYVHCtr3xX3a/dBuLRxa0J7De5S/P69Lj1/SBz/7IK7b2VTU5EjQ/kHLRBGmY3jQa4RJGETtVB+kUznovLDTDg3yHOKIZ9j6N157HB3ng8Y0SrlhYjpMXKJ8/+J8/gAAAACwVZHkAACMRHekRdCIi37n9UpwDHrOehdPXtTuB3eruFhU6VhJD3z9ARW/VtS+L+3Trgd3aeHggnbs36G5wpzmi/PK3pfViydeDH19hBM2ATDqaZQ21pfWdoaVxo7xqDGd9GcBAAAAAIgH01UBABLXnU6qm7hYn4RYn5QIOm/9f9cf77dA+fpjG697+exl5ffltefwHu39wl4VFgtaOLigtt1W/XZdmZmMDMOQ53pq2225liun4chatfTad17T6R+fHjIy2C7GvX7CuJNDaUyuAAAAAAC2BkZyAABGotd0Upv93eu8jVtQmaByN67c0K4Hdyl/f17z++e1cHBBOw/s1OzeWc0szGh6x7Ryc7lPt3xO2XxWM7tm9Parb8cRlk3165AO+nX6MItRJ90RzS/qP3Xwhc9uUa8xyHnjTDQMc5+jlEQbk/jO9rseAAAAAGw3jOQAAGxLlesVZWezys3llJ3NysyaatttWcuWXMuV790bVTJlyMyYMrOmzJypTC7T2WYyWnprSYVHC0O1I0xCY7P9/TqNw64NMUiZOKXhV/3rY9hvcfe4xd3RHbbMOJ/vJHbGxx2vYb+zQdcGAAAAgO2MkRwAgERVKpXPbGH2v/POO2t/X716NZHP5etlyZAM05Dv+XLqjuq366qWq2rcbciu2XItV57ryfd8yb833ZUpGVOdMuU3y8OEJlWS7ohOw2iCXuIYVTGo7gLWUReyDhvPUScYghZPT3PCI83v6EaT1FYAAAAASBojOQAAiblw4YIeeughnTp16teO9VocfLOpqBL77Eue48mpOWp+3FRmJiOn4cgwDLmWq9ZqS07dkWu5ajvtTsKj7XeSHjHp1UnZa5RB91ivsv3K9aszTLmo0toh22saoaQXrO6VCAj7DAZNcKQh7t37S6ukYzXsdzbstQAAAABgOyLJAQBI1Lvvvqunnnrq1/aXSqVNzy+VSp85tj5BEufnyo7O6BG7Zqu53NT0nWkZhiG7asuY6iw27tQdtVZbsuu2nMZnkx2Gaah0fPN7iEOvTu+gDs5BOsvjKBdkEjpk09y2jYZZcyWu620140xwdPfHneACAAAAgO2EJAcAYFsqLhblNBxZy5YadxvKTGfkuZ5yczmZGVO+58u1XNl1W62VluzavURHw5VrdbbCI8Otx7HVTXKHbBrbnsY2TbpJiukktRUAAAAARok1OQAA29aRJ4501uC401BtqaZqudrZKlXVlmqq36mrcbeh5idNtVb+f/buP0aOM7/v/KdquntG7BlyyCHFnh5mKGolSl4ub6VZkt5bWxa90kEXIBsgJAWvRCAKqMTI4hIIOcTAIclCYqDYyAFnYxNk14hj8XzmSl5xKSNBAm8gbFZ0ZO56V7TgrEAjWtMSvaOeofhjh+zu6uqq6q77o6dHo1b/qKr+VT3zfgEF9VQ9Tz1PP1VDAc93nudbXlvVYd+2dfCpg8PufqyFnZDttEVUXCZ4o/az235H+f6NeT+a5QCJmhMkrHarSbod06jvTFzeqSBGqa8AAAAAMGgEOQAAm9aJV0/Itdy1oEY+l1f+g1qgo7BcUPFaUdZ1S6WbJZVu1QId9m1bqkrHvnmsr33rlL+g08Rur+4XRbcTso196Vcuh24DKlH72axcp5wNQfozbEHevUGNadD3fZBj2u3vZtyfPwAAAAAMC9tVAQA2tVNvntLZJ84qv5iXa7lKppNKpBKSKfkVXxW3Is/y1lZw+L6vZy8+27P2O01wNvvr9XqdIH8dH6atVvXCWN9G2P51+m6d/iK/0/moY9KsH1H6GfTZtepf2PHsRtQxjRqQ6teYdnrmvfodajcuvfqdHeTzBwAAAIBRwkoOAMCmlj2c1cnvnFThRkEr76+sbVlVWCqsreYoXi8qv5SXW3D1zOvPKLPQ/1wc7bYRinKt0yTooLYt6qTd9xpkW0HGK8z5XrQZd+36P6wxjYuov88AAAAAgM5YyQEA2PTmjszpq+Wv6vxT5/XOt95RYiIhv+pLkgzTkGd7OvjUwcBbVAWZsOzFpGbYewxiInXUvtcw6obNq9EPUQMH3d63X/UHNaaD/n4EPwAAAACgM4IcAACsOv7KcR1/5biW315W7q2cpNpKj8xD/V+5AQAAAAAAgPAIcgAA0CDzcEaZhwlsAAAAAAAAxB1BDgAA0FKYRNJsrQMAAAAAAAaNxOMAAAAAAAAAAGAksZIDAAC0xOoMAAAAAAAQZ6zkAAAAAAAAAAAAI4kgBwAAAAAAAAAAGEkEOQAAAAAAAAAAwEgiyAEAAAAAAAAAAEYSQQ4AAAAAAAAAADCSCHIAAAAAAAAAAICRRJADAAAAAAAAAACMpMSwOwAA+IhhGGuffd8PXa5T/XbXg7YNAAAAAAAAxAUrOQAgJupBhnqAYX3QIWg53/dbBigMw1i77vv+x+q1uwYAAAAAAADEFUEOAIiReoCi00qKoOWa1Ql7DQAAAAAAAIgrtqsCAITysys/04/+3Y/03vfe060rtyRJO+7boX2/vE9H/vERTd8zPeQeAgAAAAAAYLMgyAEAm1B9e6qw117/tdf1Z7/7Z5IvTUxPaPs921WtVGVds3Tp31/S2y+9rUNfOaTHfv2xfnYfAAAAAAAAkESQAwA2nagBjt9//Pe19GdLyjyU0eTuSaXSKUmSa7sq3ynLvmXLumHpT//tn+ra29f09B893bfvAAAAAAAAAEgEOQAgVupBhk6Jv4OWa1Uv7LWzT5zV8v9Y1t6jezVz/4wmZyeVmEio4lRUvl2WddOSNWVpbGJMRsLQ1Tev6uW/9bJ0OFT3AAAAAAAAgFAIcgBATNSDFvXAxfqAw/oARKdyjZ/r1+s/ry8T5NpfnP8LLX5/UfO/NK+7P3O3dh/cra17tkqGZK/YKn5YVGIiIcM05Fd9Vd2qKk5F77/xvrRV0v7uxwYAAAAAAABohiAHAMRIq5UUjeeDluvFte999Xvavm+7ts1v045P7dCuz+zSzP4ZlW6WlM/l5Vd9VZyK3JIr13LlFB05BUfldFnud12CHAAAAAAAAOgbc9gdAADEV2GpoFt/dUtb7t6iu7bfpfHp8bVcHGbC1Nj4mBITiU8edyWUTCelW5JRDLelFgAAAAAAABAUKzkAAC3lLuXWghnGmKGqV5W9YkuGVPWq8mxPVa8q+bWtrgzTkDlmykzUDsM05OdarxIBAAAAAAAAusFKDgAYsqNHj+pLX/qSLl26tHbUnT59eqifV/5qReaYWduSqlxLMl5YLiify8v60FJ5pSy36MqzPVXcivyKL7/qrwU9xowx6Vb4MRk19RwpQRLGtyrX6tr682HrAgAAAAAAbHSG324j9h44ffq0fN/XCy+80M9mAGhj/75t5O8WZ+/+53f17V/5tuaOzGnngzs1vW9ak7snlZpKyRwzVXEqcgqO7BVb1k1L1nVLxetFFa8VVVguaOWnK/Kf9PX8N58f9lfpm/UJ3pslgw9Sbn1i+WY/N94naFkAAAAAAICNju2qAAAtZT+XVbVSVflOWaVbJSW3JOVXfaXyKZkJU37Fl1ty5eSd2lF05FlebWWHU5Ff9eVnN/4EfD3IsD6AEaZc0CBFLwIcS5eWlLuUkyRlD2U1uzDbtNzer4W67Zqrz3Wue/W5cPdcf79WdVu1GaatZvcI29cw9w5z/6h9a6w36Pb62WYvnnmze3WqH3ZMu/19GNSz6PU7GrWPYdpsda9e/d4CAAAAGA0EOQAALU3OTmrmUzMq3SipuK0oM2Gq4laUmkxpLDlW28bKqcgpOnLurB4FR67lyi250k5J6WF/i9EQJegRpt65E+d0+fxlJbckJUPSalHXcnXgyQM68eqJ0H2Oau/Xupv4DlOmfi3K5G7Q+p30q3/txjFqkKpTX3vdz6ht9vKZBR2rqGPajV6PaT/Gs1/vWrs2e9UHAAAAABsDQQ4AQFtf/I0v6vzT55WcSsowDHllby3IIUlVtyrXduUUHJXvlGXfseUUHHklT/o7Q+78COm0QqPVqo1O9RYvLurMo2eU3pnW/CPzSk2mZJiGqm51bauxK69f0YvjL+rUm6eUPZyV1HlCM8jkY7vJ1DCBjiA69TfI5G4/+9vr/gXtV/1a2Engxnv2u5/N2my8b5DyYduMoldj2knUMY1ar9vxjDIuw3qGAAAAADYWEo8DANp64G8/oHsevUe3r95WfimvfC6v/Aer/13Kq3CtIOu6pdLNkuyf2SqvlGXdtHTv4/dK9w+795vb4sVFvfyllzW7MKt9j+/T3JE5ZQ9llXkoo10/t0s77t+h6X3Tmtk/o/TutM4+cVYf/PCDvvYpyvZRUSbHw7bXqlwvJlj70b8gYxJ1Uj3MBHinckG3Ygo7/r0Y0yDtN953GBPuYcc0Sr1uxjPquAzjGQIAAADYmAhyAAA6evq/PK35X5zX7b++rUKuUAtw5PIqLBVUuFZQ8VpRxetFWTcsFW8U9anHPqUv/8cvD7vbA1PfMqpdPo525TrVi+rMo2e0474dml2YVfZQVns+v0ezn5vVrk/v0vb7tmt677S2zm3VZGZSU7NTSt6V1EuPvNSXvkS1mScugwYABr1VT2N7ce1nUHF8x6KO6ag/i6ji+AwBAAAADA7bVQEAAjn5Ryf13X/+Xb319bdUuFbQ2PiYEuO1/414tqdKuaKxu8b0C7/2Czp6+uhwOztA9STi9UBFq+2j2pXrlIi83VZVreqdO3FO6V1pzeyf0c4HdyqzkNH03mlVnIqKHxaVmEjIMAxVvaoqTkWe7cm1XNl3bL325dd07A+OdTMsXdtok7D4pGH/Bf6wg0NMzHePfycAAAAASAQ5AAAhPPavHtOhXz2kH339R3rvu+/p5k9uSpJ27N+hex+7V0f+0RFt3bN1yL0cvFYJvxvPt0sM3utrl89f1vwj80rvTmtqbkrTe6e1bX6b7BVbFaci13Ll5B2lJlO1I51SMp3UxPYJ/fjVH/ctyBEmkXgvJoE7tXf1uVqZTvkN+jUhzSTtR3oVBBj0OxZVHPoQRJwSrjcrE/fxAwAAANB/BDkAAKFs27tNj//rx4fdDbSxdGlJyS1JpSZTSm5JykyaqjgV2Su2PNuTX11dVTJmyEyYMpOmzJSpRCpROyYSWn57WZmHM131I0rS6KDXw+q0/3890DGIvgyrjSB9aBXw6edEd6t7dzuJPeh3LExboxjcikM+krDXAQAAAGwO5OQAACCipaWljx1Rzl+6dGnt+oULF3ry+Y1X3pAMyTAN+VVfbtFV8cOi8rm8rBuWnIIjz/ZU9aryq77kr+YFMSVjrFYn91auy9GJppeTv3H/S+84968e7GgX/Om1egLrbhJ8BxnTQQcYOiVyj3PAYxjvaByfIQAAAIB4YyUHAAARnD59Wvfff7+OHj36iWtBt69qPNerz7UTUtWtyi24Kt0qKTGRkGu5MgxDnu2pfKcst+jW8qm4lVrAo+LXgh490mqSstNqgV5uUxXkXu3KttvKqhtxDXC0Skzd74TVrQIBYcY/zOR4HMa9/v3iKu4Bjjg8QwAAAADxQJADAICI3n33XT399NOfOJ/NZpuWb3b+0KFDa5/XB0y6+vz0Ub33jffkFByVVkoavz4uwzDk5B0ZY7Vk427RVflOWU7RkWt9PNhhmIayh5t/h14ImgejmV5PgnYqG3aiPYhRmKSNc9+aCTum3b5jG11cAxzNyvfiXgAAAABGG0EOAAA2mNmFWbmWK3vFlnXDUmI8oapXVWoyJTNhyq/68mxPTtFR+XZZTmE10GF58uzakXmou3wcwxL3yc2496+duPY9rv0aVaMQ4AAAAACA9QhyAACwAR148oCuvH6lFtgYqyUeT02mZCZNyZcqbkWu5cop1AId9VUd9m1bB586OJQ+d7s6I+4TpWH7F3TFy7C/b9R+9mK7pihj2qt7davdd+92TKO+M6MQ4IjTMwQAAAAQDyQeBwBgAzrx6gm5lqv8Ul6F5YLyubzyH+SVz9V+Ll4ryrpuqXSzpNKtksq3y7Jv21JVOvbNY33tWz/yEHQ7udmqT73qa6/7169cDp3GoVP/o/azWbk4TsiHFeS9GtSYdqo3CgEOAAAAAGiGlRwAAGxQp948pbNPnFV+MS/XcpVMJ5VIJSRT8iu+Km5FnuWtreDwfV/PXny2Z+13mlTtdY6LTm22S24dpW5Q/exfpwBAp/Nhc6K0ErWfQce/UTdjGlXUMY0akOrXmHZ65mHHsxfjMqhnCAAAAGBjYiUHAAAbVPZwVie/c1KFGwWtvL9SW82Ry6uwVFhbzVG8XlR+KS+34OqZ159RZqH/uTiuPhevict2/YlDX9v1bZBtdWovaj/jPPbdatf/YY0pAAAAAGw0rOQAAGADmzsyp6+Wv6rzT53XO996R4mJhPyqL0kyTEOe7engUwcDb1EVZJJ0kJPvvWyvXxPAvbhv2Ht00+Yw6g7y+0W956C+2zDaHYVnPqx7AgAAAIg/ghwAAGwCx185ruOvHNfy28vKvZWTVFvpkXmo/ys3AAAAAAAA+oUgBwAAm0jm4YwyDxPYAAAAAAAAGwNBDgDAQBiGsfbZ9/3Q5drVX3+t0/V2bQNBhEkkzfY5AAAAAAD0F4nHAQB9Vw8y1AMMjUGJIOV8328boKhfbxbgWH+tVdsAAAAAAAAYPazkAAAMRD340CnQELRc2HbDWLq0pNyl1bwVh7KaXZjtuh/YOFidAQAAAABAfBDkAABsCL3YkurciXO6fP6ykluSkiFp9Tau5erAkwd04tUTPegpAAAAAAAAeoUgBwBg5LXaoqpRq/OLFxd15tEzSu9Ma/6ReaUmUzJMQ1W3KqfgyF6xdeX1K3px/EWdevOUsoezffsuAAAAAAAACI4gBwBgU2gX4Hj5Sy9rdmFWM/tnlN6dVnJLUn7Vl1twVVopybphKTWZUn4pr7NPnNXJ75wcwjcAAAAAAABAI4IcAICBqAcZOuXZCFouStvNnHn0jGYXZjW7MKudD+7U1NyUzKQpt+iqdKuk8evjSownZI6ZkqT8Yl4vPfKS9M961j0AAAAAAABERJADANB39aBFPXCxPuCwPgDRqVzj5/r1xoBIs3rNcnacO3FO6V1pzeyf0c4HdyqzkNH03mlVnIqKHxaVmEjIMAxVvaoqTkWe7cm1XNl3bDnnHIkUHQAAAAAAAENFkAMAMBCtVlI0ng9arhfXLp+/rPlH5pXendbU3JSm905r2/w22Su2Kk5FruXKyTtKTaZqRzqlZDqpie0Tci47Le8LAAAAAACAwTCH3QEAAIZh6dKSkluSSk2mlNySlJk0VXEqsldsebYnv7q6qmTMkJkwZSZNmSlTiVRCiVRCSkjGcu+21AIAAAAAAEB4BDkAAH31wAMPaGlpae2ou3btmi5cuLD286A/5y7lJEMyTKOWZLzoqvhhUflcXtYNS07BkWd7qnpV+VVf8le3vDJVC3wYppSLNCQAAAAAAADoEbarAgD0zfPPP69cLtd0yyjf9z92fiiffanqVuUWaknGExMJuZYrwzDk2Z7Kd8pyi64821PFrdQCHhW/FvQAAAAAAADA0BHkAAD0VTabbXo+k8kok8ms/Xz06NGBfl7aWltV4hQclVZKGr8+LsMw5OQdGWO1ZONu0VX5TllO0ZFrNQQ7fF9+lmAHAAAAAADAMBHkAABsSrMLs3ItV/aKLeuGpcR4QlWvqtRkSmbClF/15dmenKKj8u2ynMJqoMPyajk7PF/KdG4HAAAAAAAA/UOQAwCwaR148oCuvH6lFtgYqyUeT02mZCZNyZcqbkWu5cop1AId9VUd9m1b+sywew8AAAAAAACCHACATevEqyf04viLyi/lJUme7SmVTslMmTKM2pZVnu3JtWrbVpVvl2sBjqqk45LYrQoAAAAAAGCozGF3AACAYTr15im5BVf5xbzyubzu5O6okCsov5RXYbmg4vWirOuWSrdKsm5Z8n1fz158dtjdBgAAAAAAgAhyAAA2uezhrE5+56QKNwpaeX9F+Vwt2FFYKtSCHNeKKl4vKr+Ul1tw9czrzyizQDIOAAAAAACAOGC7KgDApjd3ZE5fLX9V5586r3e+9Y4SEwn51dpeVIZpyLM9HXzqoI5989iQewoAAAAAAID1CHIAALDq+CvHdfyV41p+e1m5t3KSais9Mg+xcgMAAAAAACCOCHIAANAg83BGmYcJbAAAAAAAAMQdQQ4AALpkGMbaZ9/3I5XrxzUAAAAAAICNjsTjAAB0oR5kqAcY1gcdgpYzDEO+768dvbgGAAAAAACwGbCSAwCALtUDF50CDa3KtVuB0evVGUuXlpS7tJpv5FBWswuzTcvt/Vq0+199rnPdq8+1vtaqbrs6Qe7R7zY76baNsN+pVb1Bt9fPNnv53Nbfq1P9sGPaze9Dq/r9eBa9fke7HZcg9+nmfQMAAACwcRDkAAAg5loFRMIES86dOKfL5y8ruSUpGZJWi7qWqwNPHmGjZ5gAACAASURBVNCJV0/0vN+t7P1a84nIdpOe9WtRJmn72WYQ/fperb5TpzbbiTKGnepGrdeubi+fW9Cxijqm3ej1mPZjPOM0LvVrBDoAAACAzYUgBwAAMVLfgmq99T+vv95YtlndxYuLOvPoGaV3pjX/yLxSkykZpqGqW5VTcGSv2Lry+hW9OP6iTr15StnDWUmdJzSDTCK2m0xtNxEZtV6Q+lHq9GLStNN4Bpl8Xn89aN/q18JORDfes9/9bNZm432DlA/bZhS9GtNOoo5p1HrdjmfYcQkSpAn7zvT72QMAAACIJ3JyAAAQE82CFN1YvLiol7/0smYXZrXv8X2aOzKn7KGsMg9ltOvndmnH/Ts0vW9aM/tnlN6d1tknzuqDH37Qs/ab6TRJGibwsV6nSdF256O2GVSQieGw9wgy+Rt1Uj3MBHincv18bkH70E37jfcdxsR52DGNUq+b8Rz0uPTiPQUAAACwsRDkAACgS/Utozol/m5XrlWAo5tk4mcePaMd9+3Q7MKssoey2vP5PZr93Kx2fXqXtt+3XdN7p7V1bqsmM5Oamp1S8q6kXnrkpcjtobeCBgAGvV1QY3tx7WdQcZwYjzqmo/4spN71Lc7fEQAAAEBvsV0VAABdqOfFqAcjWm0f1anc+v+uv94u70a7a+dOnFN6V1oz+2e088GdyixkNL13WhWnouKHRSUmEjIMQ1WvqopTkWd7ci1X9h1br335NR37g2PdD86A9Cp3BgZr2M9t2MEh3tf2GB8AAAAAQRHkAACgS622mGqXWyPI+SDXW127fP6y5h+ZV3p3WlNzU5reO61t89tkr9iqOBW5lisn7yg1maod6ZSS6aQmtk/ox6/+uG9Bjm4TYgeZ+OzVZPIgJuH5a/OP9Pq5BSkzzIn0OPQhCN5RAAAAAHFHkAMAgA1m6dKSkluSSk2mlNySlJk0VXEqsldsebYnv7q6qmTMkJkwZSZNmSlTiVSidkwktPz2sjIPZ7rqR5Sk0e3qRqmz/nw/2uyVOEx0X32uNgbNkjb3c6K72+fWSpRE5/0yyPHsl0GMV5BnPqz3FAAAAEB8kZMDAICIHnjgAS0tLa0ddevP1c9fuHBh7Xq/P7/xyhuSIRmmIb/qyy26Kn5YVD6Xl3XDklNw5Nmeql5VftWX/NWtskzJGKvVyb2VizosfRN0ArOeCLkXCZEHMbkfhwBHo/okcv0YhF48tyBjOuiJ8E4JsuM8MR/nd1QaznsKAAAAIH5YyQEAQATPP/+8crlc0+2iOp3r9+faCanqVuUWXJVulZSYSMi1XBmGIc/2VL5Tllt05dmeKm6lFvCo+LWgR4+0mhht9VfYnepGrdfuL7970WYUcZ08bpWYut8Jq7t5bnVhAhxxGPf694urOI1Vo2G9pwAAAADiiSAHAAARZbPZwOePHj06uM9PH9V733hPTsFRaaWk8evjMgxDTt6RMVZLNu4WXZXvlOUUHbnWx4Mdhmkoe7j5d+uFsJPX3dbrRj/ajPPkcV2c+9ZM2DFtNwk+Cs+n34YxBlHa3MzPCAAAAMBHCHIAALDBzC7MyrVc2Su2rBuWEuMJVb2qUpMpmQlTftWXZ3tyio7Kt8tyCquBDsuTZ9eOzEPd5eNAc6M8gR7Xvse1X6Nq1Mdz1PsPAAAAIDxycgAAsAEdePJALQfHdUuF5YLyuXztWMqrsFxQ8XpR1g1LpZ+VVL5dXlvVYd+2dfCpg8PufiijMpkZdvK109Y7cZnMjdrPXvQ7ypi2OhrL9Fu71STdjmnUd2ZY7xTbSwEAAADoBkEOAAA2oBOvnpBruWtBjXwur/wHtUBHYbmg4rWirOuWSjdLKt2qBTrs27ZUlY5981hf+9ZpAjZsvU5l2k3c9qLNILqdPG7sR78mhbsNqETtZ9jnFqZPwxTkXR/UmAZ914c5nt1uNxaH7wAAAABg8NiuCtigDMNY+9wsCXKncp3qB7m/YRht2wbQX6fePKWzT5xVfjEv13KVTCeVSCUkU/IrvipuRZ7lra3g8H1fz158tmftd5pUjRJ0aFVvfRLnsAGAqG1GuX+Qv95vPNfue3UKAHQ6H/UZNOtHlH5GfW7djGlUUcc0akCqX2Pa6ZmHHc9u3rVusPoDAAAAQB0rOYANqB6AqAcY1gckgpbzfb9t8KLT/Vu1CWBwsoezOvmdkyrcKGjl/ZW1LasKS4W11RzF60Xll/JyC66eef0ZZRb6n4uj1XZAnSZBO20j1G7St921btochKh973VbQcYqzPletBl3nd7XYYxp3PQy4fhGeGcAAAAAhGf4ff4z69OnT8v3fb3wwgv9bAaAPvp9q/+3rtWKisbzzcoFOdfq544rOXzp/Qvv673vvqfFHyxKkvZ8YY/2/fI+3fPoPdK6OAn/lgDdOf/Ueb3zrXeUmEjIr64GKE1Dnu3p4FMH+75FFQAAAAAAQD+wXRWAngq6RdVP/+Sn+vZT35a9YsswDaW2pORXfS3+YFE/+M0faGLHhH7l1V9R9uezA+g1sPEdf+W4jr9yXMtvLyv3Vk5SbaVH5qH+r9wAAAAAAADoF4IcAHomaIDjT/7vP9Ef/8s/1vS909p9cLdSkykZhiGv7MnJOyqtlFS8VtSZL57R47/+uH7+uZ8fQO+BzSHzcEaZhwlsAAAAAACAjYEgB4CeaszF0Rj4uPAvL+gHv/kD7fnCHu24b4cmM5NK3pVU1auqnC+rdKuk8RvjSt6VVOKuhL73L74n13IH/TUAoKUwCY/JDwAAAAAAQH8R5AA2qPV5MXpRLki9Trk7Vt5b0X//jf+u+S/Ma/f/slu7Pr1LW+e2yhgz5OQdWTctpdIpjSXHaver+qp6Vb1x+g3p/5A0FaqLAAAAAAAAADY4ghzABlQPPtQDEK2ShHcq1/g5SL12/vDv/qGm/8a0pvdNa2b/jDKfzWj7vdvlFBwVrhVkJAz5FV8VpyLP9uSVPLlFV86KI+s1S3om6ogAQO+wOgMAAAAAgPgwh90BAP3h+/7a0Xg+aLnGI0i9Vm05BUeLP1jU1J4pbdm5RVt2bVH67rQmtk8oNZVSMp1UaktKyS3Jj467kkpsSSi5NSn9tWR44VabAAAAAAAAANjYWMkBYCCWLi1pbHysFrgYT8iQoXKhrOK1oipORVW3WguKGJJhGjLGDJlJU2PJMY0lx2SMGap+UB321wAAAAAAAAAQI6zkADaQe+65R1/84hf17rvvrh2S9Hu/93trZYb1+T/9zn+SaZqSIVXcisr5WoAjn8ureKOo8u2yXMutBTwqVflV/2NBD9M0ZVxjJQcAAAAAAACAj7CSA9hAnnnmGeXz+U+cf/TRR4f++TNHPqPv/+H35dmenLyj0s2SxlJjcouuDNOQV66ddwqOvJL30eoOz5dfWd0aa0uw3B8AAAAAAAAANgeCHMAGMzU19Ylz99xzz9A/f/ZvflYX/6+LtQDHrZJSkyn5vi9nyqltReVV5ZU8le+UVc6X5RbdWvLxsqeKW1G1WpUxx0oOAAAAAAAAAB8hyAFgIKb3TSs1mZJ9y5Z1w9JYakxVr6pyvqyxxJj8qi+v7MktumuBDqfoyC25ckuuNC5petjfAgAAAAAAAECcEOQAMBBmwtThrxzW93/z+0pOJmWYhipORcl0UmOpMcmv5erwSp6cgqPy7bLKd8pyCo7cgiv9r5JvsF0VAAAAAAAAgI8Q5AAwMEdPH9Wf/39/rjuLdyRf8mxvLchhGKtbVpU9uZYrJ18LdJRulTQ1N6Vbv3RLIsYBAAAAAAAAYB1z2B0AsLl85Z2vaCw1ppX3V5TP5T86lvIqXCuo+GFR1nVLpZslFT4saHzruP7hn//DYXcbAAAAAAAAQAyxkgPAQKXSKf2Tv/4n+p2f/x3d+sktGaZRW82RGJPv+6o6VTmWI/nS3Qfu1t//wd+XyDcOAAAAAAAAoAmCHAAGzhgz9Ktv/ar+/P/9c138fy7q5rs3ZSZrC8uqblUzD8zoC//0C/rs3/3skHsKAAAAAAAAIM4IcgAYms/+vc/qs3+vFsi49ZNbkqQd9+8YZpcAAAAAAAAAjBCCHABigeAGAAAAAAAAgLBIPA4AAAAAAAAAAEYSQQ4AAAAAAAAAADCSCHIAAAAAAAAAAICRRJADAAAAAAAAAACMJIIcAAAAAAAAAABgJCWG3QEAAEadYRhrn33fj1Su1bX158PUAwAAAAAA2AxYyQEAQBfqQYZ6gKExKBGknGEY8n1/7WgW2KgfjfdsVw8AAAAAAGCjYyUHAABdqgcfOgUaWpUb5AqMpUtLyl3KSZKyh7KaXZhtWm7v16Ld/+pznetefa71tVZ129VpVz9IvW7bDKLX3yto3Sjj0cv2+tlmL5/b+nt1qh92TLv5fWhVvx/PYhi/e53ab3WPbscUAAAAwMZCkAMAgBiIspVVGOdOnNPl85eV3JKUDEmrt3EtVweePKATr56IdN8o9n6t+SRku4nL+rWok56d7hulzUG10ap+q3Hs1GY7nfra635GbbOXzy3oWEUd0270ekzj9LsXtG0CFgAAAAA6IcgBAEAMNObhWL/qY73Ga522qFq8uKgzj55Remda84/MKzWZkmEaqrpVOQVH9oqtK69f0YvjL+rUm6eUPZyV1HlCM8jEY7vJ1HaTl1Hrra8bZuK103ftxURr1DZarTDo53g0tjWIfjZrs/G+QcqHbTOKXo1pJ1HHNGq9bscz6ri0a58VGwAAAACCICcHAAAjrFW+DqkW4Hj5Sy9rdmFW+x7fp7kjc8oeyirzUEa7fm6Xdty/Q9P7pjWzf0bp3WmdfeKsPvjhB33tb6dJ0jCBj6B1u+lPL/SijcZy/RiPdgGsfvWzU9AsyvsQ5nuHDdoNY1I97JhGqdfNeHYzLlGfPwAAAACsR5ADAIAu1VdTdFpV0apcLxKGr1/hUXfm0TPacd8OzS7MKnsoqz2f36PZz81q16d3aft92zW9d1pb57ZqMjOpqdkpJe9K6qVHXuq6L+iNoBPAg95GqbG9uPYzqDhOpEcd01F/FgAAAAAQBdtVAQDQhfqWUfVARbttp1qVa5eIvDEA0upaY4Dj3IlzSu9Ka2b/jHY+uFOZhYym906r4lRU/LCoxERChmGo6lVVcSrybE+u5cq+Y+u1L7+mY39wLPKYAEH0KudJt+0Pq704BlcGqRfPnzEFAAAAIBHkAACga62SgTeeb5c0POg9gl67fP6y5h+ZV3p3WlNzU5reO61t89tkr9iqOBW5lisn7yg1maod6ZSS6aQmtk/ox6/+uG9Bjm4TYg9yEnMQk+D8Rf1HejVhHWRMhx1giUsfghjUO9qL5z8qYwoAAACgtwhyAACwwSxdWlJyS1KpyZSSW5Iyk6YqTkX2ii3P9uRXV1eVjBkyE6bMpCkzZSqRStSOiYSW315W5uFMV/2IkjS6Xd1hTVwOot04TMpefa427s0STPdzorvVvbudsI6S6LxfBjme/dKv8Yr6/DfCmAIAAADoDXJyAAAQ0dLS0seOsOf/8i//cu3ahQsXevY5dyknGZJhGvKrvtyiq+KHReVzeVk3LDkFR57tqepV5Vd9yV/d+sqUjLFandxbuR6MUG8NchJzEH8RHue/Oq8HO+rHINQTWPczkfX6MoPSKZF7nCfnB/mOhnn+ozymAAAAAHqPIAcAABGcPn1a3/ve9+T7/tpRt/5cu/ON13r62ZeqblVuwVXpVkmF5YLyubyK14oq3SypfKcst+jKsz1V3Eot4FHxa0GPHmmctFw/edlpEjJqvV7YzAGOVhPM3QQegrbb6lzQZx4mwBGHcY9DH9oZdICj1bkwv/NxH1MAAAAA/cF2VQAARPTuu+/q6aef/sT5bDbbtHyr85J09OjRnn1e2lpbPeIUHJVWShq/Pi7DMOTkHRljtWTjbtFV+U5ZTtGRa3082GGYhrKHW/e1W+22RepHvTA2c4BjvTj3rZmwY9pu4nwUnk+/MQYAAAAARglBDgAANpjZhVm5lit7xZZ1w1JiPKGqV1VqMiUzYcqv+vJsT07RUfl2WU5hNdBhefLs2pF5qLt8HKOIAEd7ce17XPs1qhhPAAAAAKOG7aoAANiADjx5oJaD47q1tlVVPpdXfimvwnJBxetFWTcslX5WUvl2eW1Vh33b1sGnDg67+wMXxwBHp+164jIZHbWfveh3lDFtt43a+jL91m41SbdjGvWdGfYWVd0iFwcAAACwORHkAABgAzrx6gm5lrsW1Mjn8sp/UAt0FJYLKl4ryrpuqXSzpNKtWqDDvm1LVenYN4/1tW+dJmDD1utWHAMcreq3+rlXug2oRO1ns3JxmpCPKsi7PqgxDfr7NYzxDPP8ezGmAAAAADYWtqsCAGCDOvXmKZ194qzyi3m5lqtkOqlEKiGZkl/xVXEr8ixvbQWH7/t69uKzPWs/SHLxsHXC1ms8327CNMhf1kfRTRv1PCSt6nYKAHQ6H/UZNOtHlH52qtfKIJ5buzbbnQ86Kd9Jv8a00zMPO57dvGu9eP4AAAAAwEoOAAA2qOzhrE5+56QKNwpaeX9lbcuqwlJhbTVH8XpR+aW83IKrZ15/RpmF/ufiaLUdUKcJ6UFtIxQ37SazB9lWkOcT5nwv2oy7dv0f1pjGTdjn3+2YAgAAANh4DN/3/X42cPr0afm+rxdeeKGfzQDY4Pi3BHEzau/k+afO651vvaPEREJ+tfa/fsM05NmeDj51sO9bVAEAAAAAAPQD21UBALAJHH/luI6/clzLby8r91ZOUm2lR+ah/q/cAAAAAAAA6BeCHAAAbCKZhzPKPExgAwAAAAAAbAwEOQAAA2EYxtrndjsltirXrv76a+3aMAyjbdtAEGGSHpMfAAAAAACA/iLxOACg7+pBiHqAoVlQolM53/dbBijq19qVadUmAAAAAAAARhcrOQAAA1EPPvi+3zbgELRcK81Wa9TPBb3f0qUl5S6t5q04lNXswmzofmDjYnUGAAAAAADxQZADALChhdmi6tyJc7p8/rKSW5KSIWm1mmu5OvDkAZ149UT/OgoAAAAAAIDQCHIAADaMxoBG0ADH4sVFnXn0jNI705p/ZF6pyZQM01DVrcopOLJXbF15/YpeHH9Rp948pezhbD+/BgAAAAAAAAIiyAEA2NAat6hqDHwsXlzUy196WbMLs5rZP6P07rSSW5Lyq77cgqvSSknWDUupyZTyS3mdfeKsTn7n5KC/BgAAAAAAAJogyAEAGIigeTHC5s9op1VujvXOPHpGswuzml2Y1c4Hd2pqbkpm0pRbdFW6VdL49XElxhMyx0xJUn4xr5ceeUn6Z113DwAAAAAAAF0iyAEA6Lt60KIeuGi1pVSnco2fo2xNtd65E+eU3pXWzP4Z7XxwpzILGU3vnVbFqaj4YVGJiYQMw1DVq6riVOTZnlzLlX3HlnPOkUjRAQAAAAAAMFTmsDsAANgcfN9fOxrPBy3XeLS7T6s+rHf5/GVtv2+70rvTmpqb0vTeaW2b36YtO7doYnpC41vHlZpMfXSkU0qmk5rYPiFdDvPtAQAAAAAA0A8EOQAAm9LSpSUltySVmkwpuSUpM2mq4lRkr9jybE9+dXVVyZghM2HKTJoyU6YSqYQSqYSUkIzl7rfUAgAAAAAAQHQEOQAAfbW0tPSxY/355eVlXbhwYe3cID+/8cobkiEZplFLMl50VfywqHwuL+uGJafgyLM9Vb2q/Kov+avbZJmqBT4MU8pFGREAAAAAAAD0Cjk5AAB9c/r0ad1///06evToJ67V82+s30JqkJ9rJ6SqW5VbqCUZT0wk5FquDMOQZ3sq3ynLLbrybE8Vt1ILeFT8WtADAAAAAAAAQ0eQAwDQV++++66efvrpT5zPZrOSpEwms3ZufTCk75+fPqr3vvGenIKj0kpJ49fHZRiGnLwjY6yWbNwtuirfKcspOnKthmCH78vPEuwAAAAAAAAYJoIcAIBNaXZhVq7lyl6xZd2wlBhPqOpVlZpMyUyY8qu+PNuTU3RUvl2WU1gNdFheLWeH50uZzu0AAAAAAACgfwhyAAA2rQNPHtCV16/UAhtjtcTjqcmUzKQp+VLFrci1XDmFWqCjvqrDvm1Lnxl27wEAAAAAAECQAwCwaZ149YReHH9R+aW8JMmzPaXSKZkpU4ZR27LKsz25Vm3bqvLtci3AUZV0XBK7VQEAAAAAAAyVOewOAAAwTKfePCW34Cq/mFc+l9ed3B0VcgXll/IqLBdUvF6Udd1S6VZJ1i1Lvu/r2YvPDrvbAAAAAAAAEEEOAMAmlz2c1cnvnFThRkEr768on6sFOwpLhVqQ41pRxetF5Zfycguunnn9GWUWSMYBAAAAAAAQB2xXBQDY9OaOzOmr5a/q/FPn9c633lFiIiG/WtuLyjANebang08d1LFvHhtyTwEAAAAAALAeQQ4AAFYdf+W4jr9yXMtvLyv3Vk5SbaVH5iFWbgAAAAAAAMQRQQ4AABpkHs4o8zCBDQAAAAAAgLgjyAEAQJcMw1j77Pt+V+UMw/jEtU71grYPAAAAAACw0ZB4HACALtQDDPXgwvqAQ9hyrc75vr92NJZpvA4AAAAAALCZsJIDAIAu1YMLzYIQQcvVgxWN5zutDAkb2Fi6tKTcpdV8I4eyml2YbVpu79dC3XbN1ec61736XOtrreq2q9NNvSD3i3qPVvdbL+i9m9UPUrex3qDb62ebvXzmYZ532DHt5vehVf1+PItev6Pdjks37ffidxYAAADA6CDIAQDAkEUJVqyvW9fuHudOnNPl85eV3JKUDEmrRV3L1YEnD+jEqycitR/F3q81n4RsN+lZv9bLep3q9Eq3/WtVv9U4dmqznU597XU/o7bZy2cedKx6/V5002avn0U34zmMcYljHwAAAAAMD0EOAACGKEyAo1nZ9T83u754cVFnHj2j9M605h+ZV2oyJcM0VHWrcgqO7BVbV16/ohfHX9SpN08pezgrqfOEZpAJ5HaTqe0maaPU69TfTpPC/Ra1f63+Oj3o96pfCzsJ3HjPfvezWZuN9w1SPmybUfRqTDuJOqZR63U7nmHHJUiQhhUZAAAAAIIgJwcAAENmGMbaUf+5WZmwqz0WLy7q5S+9rNmFWe17fJ/mjswpeyirzEMZ7fq5Xdpx/w5N75vWzP4ZpXendfaJs/rghx/05Du10mmSNEzgo9trzfRjcrUX/WssFyRg0Mvv3q9+dhrvXr8PYdtvvO8wJt3DjmmUet2M57DGpY6ACAAAAACCHAAAdKldcKJTufVJw9fn7GisF2U7qzOPntGO+3ZodmFW2UNZ7fn8Hs1+bla7Pr1L2+/brum909o6t1WTmUlNzU4peVdSLz3yUuh2Npq4TJYGDQAMequexvbi2s+g4vK814s6pqP+LKRofYvjMwQAAAAwOGxXBQBAF+rJwuuBi1bbR7Ur106rwMj6ezael2o5ONK70prZP6OdD+5UZiGj6b3TqjgVFT8sKjGRkGEYqnpVVZyKPNuTa7my79h67cuv6dgfHIsyHCMtzhO/G9Gw/wJ/2MEhJubbi5pcHQAAAMDmQ5ADAIAutQpYtMufEfReneq0un75/GXNPzKv9O60puamNL13Wtvmt8lesVVxKnItV07eUWoyVTvSKSXTSU1sn9CPX/1x34Ic3SbE7iavQT/u3y0maT/SqyBAkDEddoAlLn0IIq7v6KiMHwAAAID+I8gBAMAGs3RpScktSaUmU0puScpMmqo4Fdkrtjzbk19dXVUyZshMmDKTpsyUqUQqUTsmElp+e1mZhzNd9SNK0uh2dbuZzIyaKH1Q4jBRe/W52rg3SzDdz4nuVvfudhI7SqLzfhnkePbLIMYr7DOPw+8NAAAAgOEjJwcAxEhjAuqw5cLUb1WvU1185IEHHtDS0tLaUbf+XNDzP/nJT9Z+vnDhQlefc5dykiEZpiG/6sstuip+WFQ+l5d1w5JTcOTZnqpeVX7Vl/zVd8KUjLFandxbuR6MUG+FnRgOOmE6rAnnOP8lej3YUT8GoZ7AuptE1kHGdNDPu1Mi9zgHPOL6jsZ5zAAAAAAMHis5ACAm1udqqAcbmm1F1K5c/b/tAhWtrkVJbL2ZPf/888rlck3HLej2VevPr7/Wk8++VHWrcguuSrdKSkwk5FquDMOQZ3sq3ynLLbrybE8Vt1ILeFT8WtCjR1pNjLZaLdCpbpB668u260PYcr0W18njVomp+52wulUgoNfPPE7jXv9+cRWnsVovrv0CAAAAMDwEOQAgRlollI5arlE9IMJqjd7IZrN9OX/06NGuPi9tra0ScQqOSisljV8fl2EYcvKOjLFasnG36Kp8pyyn6Mi1Ph7sMExD2cPN+9oLYSevw9aLMgnabrK515OqozBJG+e+NRN2TAf5vEfRMMaAZwgAAAAgKoIcALBJtFoZsv56XbtyP7vyM/3o3/1I733vPd26ckuStOO+Hdr3y/t05B8f0fQ9073rNCKZXZiVa7myV2xZNywlxhOqelWlJlMyE6b8qi/P9uQUHZVvl+UUVgMdlifPrh2Zh7rLxzEscZ/cjHv/2olr3+Par1HFeAIAAAAYNQQ5AGAT6BTgaLzWqvzrv/a6/ux3/0zypYnpCW2/Z7uqlaqsa5Yu/ftLevult3XoK4f02K8/1vPvgHAOPHlAV16/UgtsjNUSj6cmUzKTpuRLFbci13LlFGqBjvqqDvu2rYNPHRx29yOJMjk7yK2NoiRVbrdyJS6T0VH72YvtmnqZqHrQ49nuu3c7plHfmWFv3xZEnJ4hAAAAgHgg8TgAbBKNicXDbln1+4//vt7+3beVeSij+/73+3TvY/dq7sicMg9nlPlcRpnPZjS5e1J/+m//VC//zZf78RUQwolXT8i1XOWX8iosF5TP5ZX/IK98rvZz8VpR1nVLpZsllW6VVL5dln3blqrSsW8e62vfWk1odproHOXtabrtX+N371cuh07PJmwS96D9bFYueaJwBwAAIABJREFUrhPyYQR51wc1pkF/v4Y5nnF+lgAAAADii5UcABAjQXNmhM2tEXSlRitnnzir5f+xrL1H92rm/hlNzk4qMZFQxamofLss66Yla8rS2MSYjIShq29e1ct/62XpcOAm0Aen3jyls0+cVX4xL9dylUwnlUglJFPyK74qbkWe5a2t4PB9X89efLZn7XeaVG33l+Zh6q2v0+mv44ehm/6tX+3QrG6nAECn81GfQbN+ROlnp3qtDOOZRx3TqAGpfo1pp2cedjy7edcAAAAAoBcIcgBATNSDFvXAxfogxPqgRKdyjZ+DBDMagyXr6/zF+b/Q4vcXNf9L87r7M3dr98Hd2rpnq2RI9oqt4odFJSYSMkxDftVX1a2q4lT0/hvvS1sl7Q83Duid7OGsTn7npF565CXZd2xNbJ9QIpWQMbb6rLyqPNtbW8Hx7MVnlVnofy6OIJOzYeptdK3GpR/j0U1bUesO8vsNWrt3mjGticMKEgAAAACjzfDD/ClvBKdPn5bv+3rhhRf62QyADY5/S4bn65/+usaSY9rzC3u05+f3KHskq5n9MyrdLNW2QGp2fJDXncU7Kqoo/ys8tzg4/9R5vfOtd5SYSMiv1v7Xb5iGPNvTwacO9n2LKgAAAAAAgH5gJQcAoKXCUkG3/uqW9j6yV3dtv0vj0+NKpVOSJDNhamx8TImJxCePuxJKppPSTyWjGC73B/rj+CvHdfyV41p+e1m5t3KSais9Mg/1f+UGAAAAAABAvxDkAAC0lLuUWwtmGGOGql5V9ootGVrb6qjqVSV/NbG5acgcM2UmaodhGvJzfV0wiJAyD2eUeZjABgAAAAAA2BjMYXcAADa7Cxcu6NKlSx876q5evarTp0+v/Tzozyt/tSJzzJRf9VUp15KMF5YLyufysj60VF4pyy268mxPFbciv+LXtkJaDXqMGWPSrUjDAsTW3q8FPwAAAAAAQH+RkwPASNjI/5bE+bu9+5/f1bd/5duaOzKnnQ/u1PS+aU3unlRqKiVzzFTFqcgpOLJXbFk3LVnXLRWvF1W8VlRhuaCVn67If9LX8998fthfBeiZMMELkikDAAAAANBfbFcFAGgp+7msqpWqynfKKt0qKbklKb/qK5VPyUyY8iu+3JIrJ+/UjqIjz/JqKzucivyqLz/LdlXYWAhcAAAAAAAQHwQ5AAAtTc5OauZTMyrdKKm4rSgzYariVpSaTGksOVbbxsqpyCk6cu6sHgVHruXKLbnSTknpYX8LAAAAAAAAbFQEOQAAbX3xN76o80+fV3IqKcMw5JW9tSCHJFXdqlzblVNwVL5Tln3HllNw5JU86e8MufMAAAAAAADY0AhyAADaeuBvP6B7Hr1HV//4qvxqbXuqVDqlsdSYZEh+pbaaw7VWAx23y7JuWvrU//Yp/c/7/6fEblUAAAAAAADoE3PYHQAAxN/T/+Vpzf/ivG7/9W0VcgXlc3nlc3kVlgoqXCuoeK2o4vWirBuWijeK+tRjn9KX/+OXh91tAAAAAAAAbHCs5AAABHLyj07qu//8u3rr62+pcK2gsfExJcZr/xvxbE+VckVjd43pF37tF3T09NHhdhYAAAAAAACbAkEOAEBgj/2rx3ToVw/pR1//kd777nu6+ZObkqQd+3fo3sfu1ZF/dERb92wdci8BAAAAAACwWRDkAACEsm3vNj3+rx8fdjcAAAAAAAAAcnIAAAAAAAAAAIDRRJADAAAAAAAAAACMJIIcAAAAAAAAAABgJBHkAAAAAAAAAAAAI4kgBwAAAAAAAAAAGEkEOQAAAAAAAAAAwEgiyAEAAAAAAAAAAEYSQQ4AAAAAAAAAADCSCHIAAAAAAAAAAICRRJADAAAAAAAAAACMJIIcAAAAAAAAAABgJCWG3QEAwOZgGMbaZ9/3I5XrxzUAAAAAAACMLlZyAAD6rh5kqAcY1gcdgpYzDEO+768dvbgGAAAAAACA0cZKDgDAQNQDF50CDa3KRV2BEaXe0qUl5S7lJEnZQ1nNLsw2Lbf3a5G6pKvPda579blw91x/v1Z1W7UZpq3Ge4TtZ5h7h22jWf0gdaN+p1611882e/HMm92rU/2wY9rt78OgnkWv39GofexUN+i/Tb38/QUAAACAYSHIAQAYGa2CHp0CIkGDJedOnNPl85eV3JKUDEmrRV3L1YEnD+jEqye6/AbB7f1adxPfYcrUr/Vi0jSqfvWv3ThG/U6d+trrfkZts9sxDXqvKOV6qddj2o/x7Ne71q5NAAAAANgsCHIAAEbG+gBFfRuqxs/Nfm53TZIWLy7qzKNnlN6Z1vwj80pNpmSYhqpuVU7Bkb1i68rrV/Ti+Is69eYpZQ9nJXWe0Awy+dhuMjVMoCOobturX+/HZHan8Qwy+bz+etDvFfU7Nd6z3/1s1mbjfYOUD9tmFL0a006ijmnUet2OZ5RxidJmkOAOwREAAAAAGwU5OQCMHMMw1o4o5TrV7/Y6RsvixUW9/KWXNbswq32P79PckTllD2WVeSijXT+3Szvu36HpfdOa2T+j9O60zj5xVh/88IO+9inK9lFBJsdblQkajOnnpGiQieGw9+hmTFppN9796menZxzlufbjHauXGcbkedgxjVKvm/GMOi69eoYAAAAAsJER5AAwUnqRwLqegLpVvXZJqhuvI7j6WAYJTjUrFzWo1KnemUfPaMd9OzS7MKvsoaz2fH6PZj83q12f3qXt923X9N5pbZ3bqsnMpKZmp5S8K6mXHnkpUl/6ZTNPdgYNAAx6G6XG9uLaz6Di+I5FHdNRfxbd2IjfCQAAAADYrgrAyOk2gXWQezfTbJujIG6+e1OSNLN/JnTdjaL+DBqDT9LHx7VduXZ5N6JeO3finNK70prZP6OdD+5UZiGj6b3TqjgVFT8sKjGRkGEYqnpVVZyKPNuTa7my79h67cuv6dgfHOt6bLrBhOXGN+ythYYdHIpjcGUjYFwBAAAAbCQEOQAghKAJrN/+nbd18bcu6tZf3lIiVfun1nM8zdw3oy/80y/ooVMP9b2vcdNqvBrPtxvXXl+7fP6y5h+ZV3p3WlNzU5reO61t89tkr9iqOBW5lisn7yg1maod6ZSS6aQmtk/ox6/+uG9BjjCJxHsxWTnsifROCOZ8pFdBgEG/Y1HFoQ9BxCnhOgAAAABsNgQ5AKCFZis3OiWw9sqe/sOR/6Dbi7d11/a7NHdoTmbKXEtgXbxe1H/9P/+rfvjvfqh/8MN/IGOMvB7DsnRpScktSaUmU0puScpMmqo4Fdkrtjzbk19dXVUyZshMmDKTpsyUqUQqUTsmElp+e1mZhzNd9SNK0uig18O0GfdJZCkefbz6XG38miV77uekc6t7dxsE6Nc7FsUgx7Nf4pCPpJVRCRgBAAAAQFjk5ACAJqJsTVXOl/Vv9v0blW+XtfcX9+rex+7V3OfnlHkoo7s/c7d2PrhTd3/6bm27Z5t+9t7P9FvzvyWn6PTpG8THAw88oKWlpbWjbv25TuevXr26dv3ChQs9+fzGK29IhmSYhvyqL7foqvhhUflcXtYNS07BkWd7qnpV+VVf8ldX8piSMVark3sr1+XoRNOPyd+4TijHeWK2HuyoH4NQT2DdTYLvIGM66PehUyL3uL6f0nDe0Tj/XgAAAADAoLGSA8DIqQcggiSwDpOPo7FeWN/4zDeUTCe15/N7NPPAjKayUxpLjcmzPdkrtqwbVm3FQMqUYRi6/dPb+u2Dvy09E7qpkfH8888rl8s1Hc+g21c1nuvV59oJqepW5RZclW6VlJhIyLVcGYYhz/ZUvlOWW3Tl2Z4qbqUW8Kj4taBHj7SapOy0WqCbyc1mdVu1N0xxnchtlZi63wmrWwUCwjy7MAGOOIx7/fvFFQEOAAAAABg+ghwARkovElivD3o0Xq//3Cz3RrsE1v/tX/w3le+Ude/j92rXgV3KfDajrX9jq/yqr9LNkgofFmQmzNqkej2JddnT7au3pTckPdr10MRWNpvt+vw999yz9vno0aO9+fz0Ub33jffkFByVVkoavz4uwzDk5B0ZY7Vk427RVflOWU7RkWt9PNhhmIayh5t/h17oNHndbuI3yiRo2MnyfhuFidw4962ZsGPa63dsoxmlAAfPCwAAAMBGRpADwMjpNoF11OTVra5Xvare+u23NLN/Zi159cwDM9p+73aVbpVkmEYtqGF5cou1RNblfFmpyZTGt43L/aEr45fIzTFoswuzci13bZVNYvz/b+/+YuSq7jyB/251VXfHbYONsehuEhtWTJgJ8go8EGW1YWBHo+FpHsY42kFIE0ReV0L7ONoHsJRdafYNaZ7hxUkmIPIcid0JSFkeZkA8BCGtVszADHQ7GIhNVVdX17+7D+VuOk3Xv1t/b/vzkUqqvnXPPadO3bKl861zTjHazXYsHl+MQrEQaTuNZq0Z9a167NzYiXrlZtBRbUaz1nmsPjjafhwcLs8DsvPa9nltV17lKeAAAAA46oQcACP63T//LprVZiyfWo6lE0tROlaKJEmiUW10Nq8uJJ1Nq0uFWFhciIWlhSguFaO4VIzSN0oRn0Wkvxvf8kcM7oEfPBAfvP5BJ9hY6Gw8vnh8MQqlzqybVqMVjWoj6pVO0LE7q6N2oxbnnzo/kzbnZZmhrIZ9D4POeJl1n2Rt5ziWa8rSp+O61qh6vfdR+zTrPZO3gGOel/sCAAAYBxuPA7lx+vTpKJfLe49d+zelnsXzzXc2I5KIhdJCREQ0d5pR/bwa5Y1ybH++HY2tRrTqrUjbaWcmyM3NrpOFJArFQhSSQsRs9q++5V165VI0qo0ob5ajcrUS5Y1ylD8pR3mj8/fWb7eieq0a259vx/YX27FzYydqN2oR7YiLP7k40baNe2Cy3/XmYSB01MHjg+9hUu+p23UHbX/Wdh523jwOyA+rX39GTK9PB/2e5CXg2G+e7wEAAIBRmMkB5MIzzzwTN27ciM3Nzb1jJ06ciIiIX/3qV/HMM8/M7Hn1s2okhZt7OGw3ova7WlSuVvY2sG5sd5aoam43o1VvRbvRjnarHdHuLH9VSArRrrbH3WUM6NlfPxtXnrgS5Y/L0ag2orRSiuJiMaIQkbbSaDU6S43tzuBI0zR+9NaPxlZ/v0HVcQ5MDjLw22+wvN/xcQQU/X69f9ix3TKHlZ3Ee8oSoGRtZ79y3YzSp1ll7dOsgdSk+rTfZz5sf46jX6b1GQIAAOSNkAPIhXPnznV9bTd0mNXzu/79XZG2087eDdd3YuvaViSFJOqV+t4SSPWteux8uRONrZubV9db0Wq0Im2m0Wq3Ir3LclWzsv7Iejz9y6fjpUdfitqXtVg+tRzFxWIkC0mk7TTazXY0a829GRw/eutHsXph8ntxjHvQst+yR3kfJO32/ibxvkapK2vZab6/aet1b+rT0eRhJg8AAMCokrTfLrsjunz5cqRpGi+88MIkqwGYmXqlHn976m/jzHfOxKl/dypu++ZtcezMsa82sG51NrDeKe/Ezo2d2P5iO6qfV6N6rRpbn25F+Wo54m8inv/x87N+K7e81556Ld77+XtRXC5G2u7895gUkmjWmnH+qfMTX6IKAAAAgOGYyQEwosXji/Gt//Ct+Pz/fR5LJ5aiUCxEq9GKxZV9G1jXOxtY7wYd9XI96lv1qFfqEWcj0qKZHPPgyZ89GU/+7Mm4+u7V2Hi7s1HK+iPrsfrg5GduAAAAADA8IQfAGPzllb+Mv/v230Xl00pE0tl8fHFlMRYWO5uR7y55tLts1W7Q0aw1I56cceP5mtWHVmP1IcEGAAAAwLwTcgCMwe1nb48/ef5P4tf/49eRttNobDe+CjmSmxtY11udTcgrN4OO8k786X//03h96/UIEzk4YobZSNp+AQAAAEBWQg6AMXn0bx6N0nIp/uG//UPUy/VYOrEUC0sLkRSSSNM02o2bsznK9WjUGvHn//PP45H/8ki8fvn1WTcdAAAAAHJJyAEwRt/7r9+Ls//xbLz6n1+NytVKtJqtWCh1lqxq1VuxsLgQK3euxF//r7+O1QuWQ+LoMjsDAAAAmAYhB8CYrX93PZ775+fiX//Pv8a//O9/iX97698iIuJb3/9W3Puf7o2z3z874xYCAAAAwNEg5ACYhCTi7PfPCjQAAAAAYIIKs24AAAAAAABAFkIOAAAAAAAgl4QcAAAAAABALgk5AAAAAACAXBJyAAAAAAAAuSTkAAAAAAAAcknIAQAAAAAA5JKQAwAAAAAAyCUhBwAAAAAAkEtCDgAAAAAAIJeEHAAAAAAAQC4JOQAAAAAAgFwScgAAAAAAALkk5AAAAAAAAHJJyAEAAAAAAOSSkAMAAAAAAMil4qwbAMBXkiTZe56m6dDn7T/e67XDrj1o3QAAAAAwL8zkAJgTuyHDbsBwMLAY9Lw0Tfcehx3vds395brVDQAAAADzxEwOgDmyG0L0CxoGPW+SNt/ZjI13NiIiYv3h9Vi7sHboeedezHb9j57rX/aj54a75v7rDVN20HLjbu8wdQx67cPKD1L2YLlp1zfJOkft027X6ld+2D4d9f6a1mcx7ns0axtHqXOYsgAAAMyWkAPgiMmy7NTBsKRXuVcvvRrvv/Z+lI6VIpKIuHlqo9qIB37wQFx65VKmdmdx7sXRBjEnWW4SerVl97Usg8+9+nES/dbvc8vSzqx1jtqng14ry3njNO4+nUR/Tupey1rnsJ8/AAAAsyHkADhCDoYTu8tQ9XPwvMPKffzWx/HyYy/Hyp0rcfbRs7F4fDGSQhLtRjvqlXrUrtfig9c/iB8v/Tie/fWzsf7IekT0H1wcZACx12DqMEHHtEyyPf36c5DB5/2vD9qPu68NOxB98JqTbudhdR687iDnD1tnFuPq036y9mnWcqP25zj7pVed/WbdzPO/MQAAAHzFnhwA9PXxWx/HT//ip7F2YS3u/bN74+7v3h3rD6/H6oOrceaPzsQdf3BHnLz3ZJz+9ulYuWslrjxxJT75x08m2qYsy01lHRyelwHOQQaGh73GIIHBOPttUu3s91kNe3yQOoep/+B1Z3FPDdunWcqN0p9Z+6VXuawh6qBlAQAAmD0hB8Ac2V0yqt8+G4OeNy4vP/Zy3HHfHbF2YS3WH16Pb37vm7H2x2tx5jtn4tR9p+LkuZNx2923xfHV43Fi7USUvlGKlx59aSptG1TWAcujMNA5aAAw7WWUDtY3r+0c1DzeK1n7NO+fBQAAALcOy1UBzIndfTF2g4tuy0f1O+/gNQ977WDZXntyvHrp1Vg5sxKnv3067vzDO2P1wmqcPHcyWvVWbH26FcXlYiRJEu1mO1r1VjRrzWhUG1H7sha/+KtfxMW/vzhax4xoFntxZN2km2xmPeNm1uGQ+wsAAIBbmZkcAHMkTdO9x8Hjg5436GuDXvP9196PU/edipW7VuLE3Sfi5LmTcfvZ2+PYncdi+eRyLN22FIvHF796rCxGaaUUy6eW4zev/GbULulqkIHleVmm6tyLkx8I94v6r+z296j9Psl7bJymcX+Nwyw3XO+1dFqvzdW7lQUAAGB+mMkBQFeb72xG6VgpFo8vRulYKQqlQrTqrahdr0Wz1oy0fXNWyUIShWIhCqVCFBYLUVwsdh7Lxbj67tVYfWh1pHZk2TR60NcnXW7aA7vzMCD70XNfDbxPsz8mNVg9qXssi1nfX+Mwyf46rD/67RPSKySah+8TAAAAvZnJATAH7r///tjc3Nx77Np/bJTjb775ZqbnG+9sRCQRSSGJtJ1GY6sRW59uRXmjHNXPqlGv1KNZa0a72Y60nUakN5fCKkQkC50yG29vjKmXhjOLZar6bbQ9qQHpef7F+bhmVQxjdyPqUTb4HqRP5yHAmsb9NQ6zvEfnuV8AAAAYnZkcADP2/PPPx8bGxteWiYr4+jJV4zg+7PNII9qNdjQqjdj+YjuKy8VoVBuRJEk0a83Y+XInGluNaNaa0Wq0OoFHK+2EHmPSbWC032yBWS9TtWv31+KTMK8BR7elgCa9YXW3IKDbvXKYYQKOeej3Sd5f4zDNvjqsjl6ffa+2DXPPAAAAMDtCDoA5sL6+PtHjjz/+eKbn6w93rlev1GP7+nYsXVuKJEmiXq5HstDZbLyx1YidL3eivlWPRvX3w46kkMT6I4e3aRz6DV73GvjtN7iZpdy0zVNbupnnth1m2D7Ny70yK/PQB93+nejXtmHDMQAAAGZDyAFAV2sX1qJRbUTtei2qn1WjuFSMdrMdi8cXo1AsRNpOo1lrRn2rHjs3dqJeuRl0VJvRrHUeqw+Oth8Hh5uHweOs5rXt89quvNKfAAAATIOQA4CeHvjBA/HB6x90go2Fzsbji8cXo1AqRKQRrUYrGtVG1CudoGN3VkftRi3OP3V+Jm3OuszQpJYnGvdSQsO2ZdAZL7MejM7aznEs15SlT8d1rVH1eu+j9mnWe2Ze7ikAAACOPhuPA9DTpVcuRaPaiPJmOSpXK1HeKEf5k3KUNzp/b/12K6rXqrH9+XZsf7EdOzd2onajFtGOuPiTixNt2zztQ9CtLfuPj2PAd9TB44PtnPReId2O92t/1nYedt5RGJAfx/01rj7tV26eQp5RXp+nf18AAADoLkm77VI7JpcvX440TeOFF16YZDUATNDGP23ElSeuROkbpVi+YzlKK6UoLhYjChFpK41WoxXNanNvBkeapvHD138Yqxe6L1U1zObO/QwzmDqpTckHaeuog76j9ke/X/yPs85+5bJ+7qPcL8PuvzJovd2uOc4+HfX+mkSfjrs/J3WvjVrnPIdfAAAAmMkBwADWH1mPp3/5dFQ+q8T1D693ZnNslKOyWdmbzbF1bSvKm+VoVBp9A45x+ei5+RmA7NWOeWlnrw2Wp1lXv/qytnOUOufdqPfXJPp0XgzyHrLcG0fhvgEAALgVmMkBwFBee+q1eO/n70VxuRhpu/NfSFJIollrxvmnzk98iSoAAAAA2GXjcQCG8uTPnownf/ZkXH33amy8vRERnZkeqw9OfuYGAAAAAOwn5AAgk9WHVmP1IcEGAAAAALMj5ACAGRl0w+QIewMAAAAAHMbG4wAAAAAAQC6ZyQEAM2J2BgAAAMBozOQAAAAAAAByScgBAAAAAADkkpADAAAAAADIJSEHAAAAAACQS0IOAAAAAAAgl4QcAAAAAABALgk5AAAAAACAXBJyAAAAAAAAuSTkAAAAAAAAcknIAQAAAAAA5JKQAwAAAAAAyCUhBwAAAAAAkEtCDgAAAAAAIJeEHAAAAAAAQC4JOQAAAAAAgFwScgAAAAAAALkk5AAAAAAAAHJJyAEAAAAAAORScZqVJUmy9zxN08znHfb6INcetH4AAAAAAGD+TW0mx27AcFgoMcx5SZJEmqZ7j10H/z7suoeVAwAAAAAA8mmqMzl2w4U0TbuGHL3O2w0qhpWl3OY7m7HxzkZERKw/vB5rF9YOPe/ci0M3JyIiPnquf9mPnut/ncOu0atctzoHqWsYo9Yz7PvqVm7a9U2qznF/bvuv1+8aw/bpqPf1tD6Lcd+jk77XRqkTAAAAAI6qqYYc45B1yalBy7166dV4/7X3o3SsFJFExM1TG9VGPPCDB+LSK5eGbnNW514cftB7/2sHy2Ypk8Wo9XQrn7U/eunX1nG3M0ud4/7cBu2rrH06inH3abdyo/TppO61SdQJAAAAAEdd7kKO/QHFMDM0+pX7+K2P4+XHXo6VO1fi7KNnY/H4YiSFJNqNdtQr9ahdr8UHr38QP176cTz762dj/ZH1iOg/KDnI4HOvQdh+g97Dlu3X3n6DyYPKWk+39zZo+3ZfG3ZQuFsfT6qdh9V58Lr9zh22vqzG1af9ZO3TrOVG7dNh+mWU72zWOgEAAADgVjC1PTnm2cdvfRw//YufxtqFtbj3z+6Nu797d6w/vB6rD67GmT86E3f8wR1x8t6Tcfrbp2PlrpW48sSV+OQfP5lom7KGI1mOD1rfoMZRz6DhzP7Xsw6qDzMA3u+8QZdiGuYzGufnNmz4NovlkIbt0yzlRunTUfoly3dz1DoBAAAA4Cibasixu2RUr/04hjlvXF5+7OW44747Yu3CWqw/vB7f/N43Y+2P1+LMd87EqftOxclzJ+O2u2+L46vH48TaiSh9oxQvPfrSVNp2Kxk0AJj2r9gP1jev7RzUPA6WZ+3TvH8WAAAAAMBoprZc1e4m4rvBRbflo3qdd3Aj8oPXOPi82wbm+8u9eunVWDmzEqe/fTru/MM7Y/XCapw8dzJa9VZsfboVxeViJEkS7WY7WvVWNGvNaFQbUfuyFr/4q1/Exb+/OHrncEsY574no9Q/q/rmMVwBAAAAAPJtqjM50jTdexw8Psh5/a5x8DFIufdfez9O3XcqVu5aiRN3n4iT507G7Wdvj2N3Hovlk8uxdNtSLB5f/OqxshillVIsn1qO37zym1G6o6deA9L9fp0+yc2oR+UX9V859+LvP7JeY5jzZhk0jPI+p2kSbZzEdxYAAAAAyOHG4+O0+c5mlI6VYvH4YpSOlaJQKkSr3ora9Vo0a81I2zdnlSwkUSgWolAqRGGxEMXFYuexXIyr716N1YdWR2rHIIHGYcd7DRqPe8+AcZuHwdz9fdhrc/dxm8RA9yh7uEzCNPtzUsbdX5P6zgIAAADArWwqIcf9998fm5ube3+vra1FRPzesXEe/+STT+Lhhx+OiIg333wzHnvssUOfH/+/xyOSiKSQRNpOo7HViK1Pt6JVb0XaTqNeqUez1ox2sx1pO41Iby6FVYhIFjplNt7eGDnkmLVp/Yp8nn+tPotB+FGDgEH7c9rvrdvm6bvtOCxUmhfzfI8CAAAAAF838ZDj+eefj42Nja5LTx1m1OMH9/Ho+TyNaDfa0ag0YvuL7SguF6NRbUSSJNGsNWPny51obDWiWWtGq9HqBB6ttBN6jEm3AdVuswx2X+tWtle5Ya4zTvM6eNxtGaFJb1iziX9GAAAGEElEQVTdKwgY5LMbNuCYh37fH3TMo0n31bi+swAAAADAV6Yyk2N9fX2qx/f//fjjj3d9vnlbZwZIvVKP7evbsXRtKZIkiXq5HslCZ7PxxlYjdr7cifpWPRrV3w87kkIS648c3qZx6Dbo3W8wdtDB8ls94Nhvntt20Lj3XMnD5zNpsww4do8LOgAAAABgeLf0nhxrF9aiUW1E7Xotqp9Vo7hUjHazHYvHF6NQLETaTqNZa0Z9qx47N3aiXrkZdFSb0ax1HqsP5nOpKgFHf/PY9nlsU97pUwAAAADIr8KsGzBrD/zggShvlKN6rRqVq5Uob5Q7j81yVK5WYuvaVlQ/q8b277Zj58bO3qyO2o1anH/q/Kybn8m8Bhz9loial8HorO0ctd1Z3v9Hz3V/HDxn0nrNJhm1T7PeM/NyTwEAAAAA2dzyIcelVy5Fo9rYCzXKG+Uof9IJOipXK7H1262oXqvG9ufbsf1FJ+io3ahFtCMu/uTiRNvWb/+CfgO7vV6bt4CjW/luf4/LqIFK1nYedl6/PRsGac+sDXJPTqtPB/3+TLNPs3xnAQAAAIDukrTbbt63kI1/2ogrT1yJ0jdKsXzHcpRWSlFcLEYUItJWGq1GK5rV5t4MjjRN44ev/zBWL3RfqmqQAdRBBzZ7DXoPU26U+oYxaj2D/OJ/XHX2K5f18xvlc5/W5zboLIdh6s1yXw5a5zjLTfq7d1jZUfpmWt9dAAAAAMibW34mR0TE+iPr8fQvn47KZ5W4/uH1vSWrKpuVvdkcW9e2orxZjkal0TfgGJdeywhlfS0Per2vadbVr76s7Tyqn1tE7/bPqk/nxVH+zgIAAADArJjJccBrT70W7/38vSguFyNtd7omKSTRrDXj/FPnJ75EFQAAAAAAMBghRxdX370aG29vRERnpsfqg5OfuQEAAAAAAAxOyAEAAAAAAORScdYNYP4NuulxhH0FAAAAAACYHhuPAwAAAAAAuWS5KgAAAAAAIJfM5AAAAAAAAHJJyAEAAAAAAOSSkAMAAAAAAMglIQcAAAAAAJBLQg4AAAAAACCXhBwAAAAAAEAuCTkAAAAAAIBcEnIAAAAAAAC5VHjzzTfjjTfemHU7RnZU3gcAAAAAADCYwhtvvHEkwoGj8j4AAAAAAIDBWK4KAAAAAADIJSEHAAAAAACQS0IOAAAAAAAgl4offvhhfPjhh3H58uVZt2Ukb7zxRtxzzz2zbgYAAAAAADAlxYiINE0jTdNZt2VkR+E9AAAAAAAAgynec889ce7cuXjhhRdm3ZaRXL58WcgBAAAAAAC3EHtyAAAAAAAAuSTkAAAAAAAAcknIAQAAAAAA5FLx8ccfPxJ7WRyV9wEAAAAAAAwmSSUDAAAAAABADlmuCgAAAAAAyKXi/j+SJPm9F3tN8th/7mHn9bvWOF/PUj8AAAAAAJBvxYMHdsOAgyFBlvO6nbP7d6/XDws19h/rVf/Bc5MkOfSaAAAAAABAfu0tV3VYiNAv6OhmkGv1en3UMOJgeeEGAAAAAAAcPfbkAAAAAAAAculry1VNy/7ZHt1mjIxrdoelqgAAAAAA4OiZyUyO/eFGrwAiTdO9x7iWzgIAAAAAAI6Gmc3kmEbwIOAAAAAAAICja28mx8HZEgcDgmFmUgx7rVHq6kbAAQAAAAAAR9vXZnIMGjAcDDEivj47o9u1DoYgB8sdtjxVr2sfrH/3737XAAAAAAAA8itJjfwDAAAAAAA5NJONxwEAAAAAAEYl5AAAAAAAAHJJyAEAAAAAAOSSkAMAAAAAAMglIQcAAAAAAJBLQg4AAAAAACCXhBwAAAAAAEAuCTkAAAAAAIBcEnIAAAAAAAC5JOQAAAAAAAByScgBAAAAAADkkpADAAAAAADIJSEHAAAAAACQS0IOAAAAAAAgl4QcAAAAAABALgk5AAAAAACAXBJyAAAAAAAAuSTkAAAAAAAAcknIAQAAAAAA5JKQAwAAAAAAyCUhBwAAAAAAkEtCDgAAAAAAIJeEHAAAAAAAQC4JOQAAAAAAgFwScgAAAAAAALkk5AAAAAAAAHJJyAEAAAAAAOTS/wdm8ywpbdYncAAAAABJRU5ErkJggg==)
This phylogenetic tree was assembled by converting VCF data into phylip format with the vcf2phylip program and performing Maximum-likelihood based phylogenetic inference with RAxML. Only SNP sites that had data present for >= 80% of the population were included in this analysis. The ete3 toolkit was used to plot the resulting newick tree file. Each node represents a genetic division in the population and the edge/branch length (noted on each branch) corresponds to the accumulated genetic distance. All data used to produce this phylogeny and the 400 dpi png file can be found in ./VCF/Phylogeny.
6 Genome Imputation
Imputation of Genomic Sites:In addition to the variants discovered by the pipeline, we are providing you with a fully phased and imputed variant call set (./VCF/Imputed_Sites/SNPs.mergedAll.MAFgt0.05.imputed.vcf.gz) that was generated via full genome imputation with the Beagle algorithm (Browning et al. 2007 & Browning et al. 2016). Genotype imputation allows users to accurately fill in the gaps in genotype data inherent to GBS variant calling. Although imputation algorithms can very accurately phase and impute sites, the accuracy of the algorithm is highly dependent on population specific parameters including: genetic relatedness, the length of Identical By Descent (IBD) haplotype blocks, predicted recombination rates and the number of nearby markers considered during imputation. If the population is closely related the IBD haplotype blocks will be larger than if the population is diverse. Imputation algorithms move along each contig looking for sections of shared haplotype in the population and fill in the missing sites with either the variant identified to have a similar haplotype in a reference panel or the highest probability variant based on the population haplotype data and the algorithms statistical model. Before using the imputed markerset for any downstream analyses users are highly encouraged to:
- Read the literature regarding best practices for genotype imputation in their organism of study.
- Evaluate the parameters within ./VCF/Imputed_Sites/SNPs.mergedAll.imputed.log to ensure they conform to the proper standards.
- Perform validation of the imputed SNP sites against a known reference panel.
7 Interpreting Results
Analysis and interpretation of genomic data generated by sequencing technologies are among the most complex problems in genomic sciences. One consequence of this reality is that analysis and interpretation of sequencing data is far from routine. Analytic “best practices” remain very dynamic and reflect our current limited understanding of genomics including technical aspects of data collection.
The process the UW-BRC pipeline uses to analyze your GBS data can be stated as the following:
After VCFs are output from the pipeline, we perform filtering, imputation and generate population analyses (PCA and Phylogenetic Trees) to provide you with insight into the genetic relationships in your GBS population
7.1 Did it work?
Due to the many possible reasons a scientist might have for performing GBS we cannot a priori determine if your experiment was successful for your purposes. However, several metrics may be of interest to you. The first of which is the number of input reads (BarcodedReads) detected per sample. Although no clear rules exist, for a sizable eukaryotic genome (>1 Gb), samples which have in excess of 1x106 reads and low library duplication levels will tend to have good representation of the genotyped sites. Another metric of interest is the total number of SNPs called, and the chromosomes/contigs they are detected on. If you have less SNP sites than you might like on a specific chromosome/contig you should consider the following question. Was the area I wanted to cover reasonably well represented by the enzyme cut site I used in GBS? If not then trying a different enzyme or enzyme combination to cut your genome may yield better representation of that region. If the genome you are using had many contigs and you don't see them in the list, do not worry, for visualization purposes we have only included the top 30 contigs (all SNPs from the missing contigs are still present in your VCFs).
To evaluate how confident you can be in the results, you must go beyond the variant list itself, and evaluate additional aspects of the analyses and perform some basic checks.
8 Credits
8.1 Bioinformatics Resource Center

Bioinformatics Resource Center
Genetics & Biotechnology Center, Room 2130
425 Henry Mall, Madison WI 53706
Email: brc@biotech.wisc.edu
Website: https://bioinformatics.biotech.wisc.edu
8.2 Software
Platform: x86_64-pc-linux-gnu (64-bit)
8.3 References
Browning, S., Browning, B. (2007). Rapid and accurate haplotype phasing and missing data inference for whole genome association studies by use of localized haplotype clustering. Am J Hum Genet, 81, 1084-1097.
Browning, B., Browning, S. (2016). Genotype imputation with millions of reference samples. Am J Hum Genet, 98, 116-126.
Catchen, J., Amores, A., Hohenlohe, P., Cresko, W., Postlethwait, J. (2011) Stacks: Building and Genotyping Loci De Novo From Short-Read Sequences. G3, 1(3),171-182.
Ewing, B., Hillier, L., Wendl, M. C., & Green, P. (1998). Base-Calling of Automated Sequencer Traces Using Phred. I. Accuracy Assessment. Genome Research, 8(3), 175–185. https://doi.org/10.1101/gr.8.3.175
Ewing, B., & Green, P. (1998). Base-calling of automated sequencer traces using phred. II. Error probabilities. Genome Research, 8(3), 186–194. https://doi.org/10.1101/gr.8.3.186
Glaubitz, C., Casstevens, T., Liu, F., Elshire, R., Sun, Q., & Buckler, E. (2014) TASSEL-GBS: A High Capacity Genotyping by Sequencing Analysis Pipeline. Plos One, 9(2):e90346. https://journals.plos.org/plosone/article?id=10.1371/journal.pone.0090346
Jiang, H., Lei, R., Ding, S. W., & Zhu, S. (2014). Skewer: A fast and accurate adapter trimmer for next-generation sequencing paired-end reads. BMC Bioinformatics, 15(1), 1–12. https://doi.org/10.1186/1471-2105-15-182
Langmead, B., & Salzberg, S. (2012). Fast gapped-read alignment with Bowtie 2. Nature Methods, 9 357–359
Li H. (2013). Aligning sequence reads, clone sequences and assembly contigs with BWA-MEM. arXiv:1303.3997.
Li, H., Handsaker, B., et al. (2009). The Sequence Alignment/Map format and SAMtools. Bioinformatics (Oxford, England), 25 (16), 2078–9. https://doi.org/10.1093/bioinformatics/btp352